Synovial cell sarcoma is one of the most common soft-tissue tumors in adolescents and young patients, with approximately one third of cases occurring in the first two decades of life. Mean age of patients at diagnosis is approximately 30 years.
Prognosis of this relatively rare tumor is related to initial care. Survival rates have improved in the past 20 years because of treatment with primary radical surgery, along with chemotherapy and radiation.[1, 2, 3, 4]
NextThe origin of synovial cell sarcoma is unclear. Its name notwithstanding, synovial cell sarcoma is not associated with synovial joints. The basis for the name synovial cell sarcoma is the similarity between cells of this tumor and primitive synoviocytes.
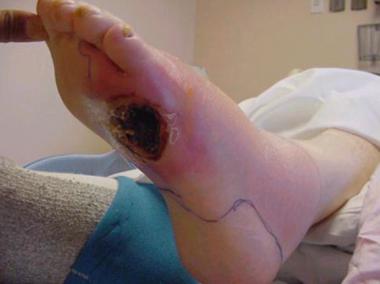
A neurologic origin for this sarcoma has been suggested. In fact, there is a histologic resemblance between neural cells of malignant peripherical nerve sheath tumor (MPNST) and synovial cell sarcoma.[5] Typically, synovial cell sarcoma is associated with a history of a long-standing nodule, sometimes present for years, which increases rapidly in size over a few months; therefore, it is sometimes overlooked. The tumor spreads along fascial planes and thus can be much more widespread than it appears on initial evaluation.
The (X;18)(p11;q11) translocation fuses the SYT gene from chromosome 18 to a homologous gene at Xp11 (SSX1, SSX2, or SSX4). The fusion proteins SYT-SSX1 and SYT-SSX2 are believed to function as aberrant transcriptional regulators, resulting in either activation of proto-oncogenes or inhibition of tumor suppressor genes.
A correlation appears to exist between the histologic subtype of the tumor and either of these two fusion proteins. Biphasic tumors, containing both epithelial and spindle cell components, express the SYT-SSX1 transcript, whereas monophasic tumors with only a spindle cell component may express either transcript.[5, 6, 7, 8, 9]
Synovial cell sarcoma is characterized by a specific chromosomal translocation, t(X;18)(p11;q11).[10] This defect appears to be the underlying cause of the tumor. This specific chromosomal translocation between chromosome X and chromosome 18 has been noted in more than 90% of cases. This fusion gene is called, in genetic terms, the SYT-SSX1, SYT-SSX2, or SYT-SSX4. These terms correspond to a fusion of the SYT gene (chromosome 18) with the SSX gene (chromosome X).
Females are more commonly affected than males in both SYT-SSX2 and SYT-SSX1 types. This association is stronger in SYT-SSX2. To our knowledge, the origin of this translocation has not been identified.[5, 6, 7, 8, 9, 11]
The incidence of synovial cell sarcoma has been estimated to be 2.75 per 100,000.[12] The majority of cases involve the lower extremities. Approximately 800 new cases occur in the United States each year, and this tumor represents around 5-10% of all soft-tissue sarcomas. Synovial cell sarcoma is the third most common soft-tissue tumor in adolescents and young adults.[13]
Synovial cell sarcoma has been reported to be particularly likely to metastasize.[14] Many factors modify patient outcome,[8, 15, 16, 17] such as tumor size,[18] anatomic localization, and histologic grade. Nevertheless, histologic criteria such as nuclear grade, measures of mitotic count, and amount of necrosis are subjective and sometimes difficult to compare.
Survival analysis is correlated with the location of the tumor in three anatomic regions:
Distal-extremity tumors have a better prognosis than proximal-extremity or truncal tumors do.[12] Nevertheless, this malignancy can affect any part of the appendicular skeleton.
Synovial cell sarcoma has survival rates of 50-60% at 5 years and 40-50% at 10 years. However, advances in oncologic therapy, particularly the development of monoclonal antibodies, may improve survival rates.
A slight improvement in survival rate has been reported with the use of chemotherapy as adjuvant therapy.[13, 2, 3] Recurrence has been reported up to 69 months after treatment and suggests a worse prognosis with low survival rates. Distant metastases at presentation suggests a bad prognosis (2-year survival rate of 25%).
Campbell et al described prognostic factors that correlated with a better prognosis, including the following[20] :
Crowson et al conducted a retrospective review of medical records to determine clinical and pathologic factors affecting survival in primary synovial cell sarcoma of the head and neck.[21] Their study included 28 patients who underwent surgery for the removal of the primary lesion, of whom nine received adjuvant radiation therapy, two received chemotherapy, and 14 received postoperative chemoradiation therapy.
The investigators found that the presence of metastases on initial presentation and tumor size exceeding 4 cm were factors associated with decreased survival.[21] They concluded that adding chemotherapy to postoperative radiotherapy does not increase survival or disease control.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved