In 1903, Batten described 3 children who had proximal muscle weakness from birth. Biopsy of their muscles showed evidence of chronic myopathy without distinguishing characteristics. In 1908, Howard coined the term congenital muscular dystrophy (CMD) when he described another infant with similar features. Ullrich first described the combination of joint hyperlaxity and proximal contractures in 1930 in the German literature; this was the first case of what is now known as Ullrich congenital muscular dystrophy.
In 1960, Fukuyama et al described a common congenital muscular dystrophy in Japan that always had features of muscular dystrophy and brain pathology.[1] Other diseases involving the muscle, eye, and brain were subsequently described: a Finnish variant (originally called muscle-eye-brain disease and Walker-Warburg syndrome. As has become clear with molecular genetics, all of these CMDs are likely caused by a similar molecular pathologic event, abnormal glycosylation of α-dystroglycan.
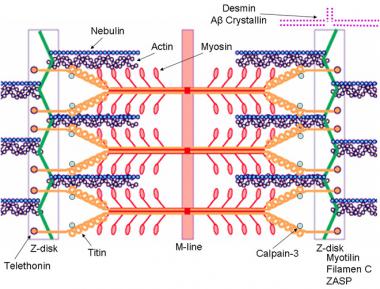
In general, CMDs are autosomal recessive diseases resulting in severe proximal weakness at birth (or within the first 12 mo of life) that is either slowly progressive or nonprogressive. Contractures are common, and CNS abnormalities can occur. Muscle biopsy shows signs of dystrophy, including a marked increase in endomysial and perimysial connective tissue; variability in fiber size with small, round fibers; immature muscle fibers; and (uncommonly) necrotic muscle fibers. No distinguishing features are present in muscle biopsy specimens, differentiating these disorders from the congenital myopathies.
Several authors of review articles have proposed classifications for the congenital muscular dystrophies. Recent classification schemes follow the following pattern[2, 3] :
Defects of structural proteins
Defects of glycosylation
Proteins of the endoplasmic reticulum and nucleus
Mitochondrial membrane protein
The OMIM classification of defects of glycosylation is as follows:
Only the muscular dystrophies with known genetic mutations are discussed in more detail later in this article. Several rare forms of congenital muscular dystrophy are not discussed in this article because of the lack of precise molecular and/or genetic information. The diagnosis of congenital muscular dystrophy is now based on clinical findings, muscle biopsy results, and genetic information.
NextThe pathophysiology of the congenital muscular dystrophies depends on the specific genetic defect for each of the dystrophies and is discussed with each of the congenital muscular dystrophies below.
International
An Italian study identified mutations in 220 of 336 patients (65.5%). The most common forms of CMD were those with α-dystroglycan glycosylation deficiency (40.18%) followed by those with laminin α2 deficiency (24.11%) and collagen VI deficiency (20.24%). The forms of CMD dystrophy related to mutations in SEPN1 and LMNA were less frequent (6.25% and 5.95%, respectively).[4]
In Japan, Fukuyama congenital muscular dystrophy is fairly common. It is approximately 50% as common as Duchenne muscular dystrophy. The estimated prevalence is approximately 7-12 cases per 100,000 children. In Italy, the prevalence of all congenital muscular dystrophies has been estimated to be 4.7 cases per 100,000 children, while in Sweden the incidence is estimated at 6.3 cases per 100,000 births. Only about 25-50% of patients with CMD have an identifiable genetic mutation.[2]
Morbidity and mortality rates depend on the type of congenital muscular dystrophy.
The major causes of morbidity and mortality are related to respiratory insufficiency, bulbar and limb weakness, contractures, seizures, ocular pathology, and mental retardation and associated brain pathology.
Some children die in infancy, whereas others can live into adulthood with only minimal disability.
These autosomal recessive diseases affect both sexes equally.
Patients with congenital muscular dystrophy present at birth or within the first year of life.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved