Transthyretin (TTR) is a protein that functions as a transporter of thyroxine and retinol and is produced chiefly by the liver (>95%), with additional production within the choroid plexus of the brain and the retinal pigment epithelium. However, it is also associated with the formation of amyloid fibrils, leading to TTR-related amyloidosis (ATTR), in which these fibril proteins are deposited into various organs and tissues, preferentially the nervous system and cardiac tissue, resulting in their inherent dysfunction.
The presenting signs and symptoms in patients with ATTR are fairly nonspecific and often attributed to more common diseases affecting both the heart and the peripheral and autonomic nervous sytems.
Patients with cardiac deposition typically present with the following typical symptoms of chronic heart failure (CHF):
Neuropathic involvement in patients affected by ATTR–familial amyloid polyneuropathy (FAP) is classically a symmetric, ascending length−dependent, sensorimotor, axonal polyneuropathy subtype and may include the following:
Carpal ligament deposits: Weakness and paresthesias of one or both hands (eg, variant TTR L58H, normal-sequence TTR); localized symptomatic carpal ligament deposition sometimes precedes other clinical manifestations by as long as 20 years
Patients with rare TTR variants that cause CNS disease may present with the following features:
See Clinical Presentation for more detail.
Physical examination findings in patients with ATTR depend on the organ involved, which is affected by the presence and genetic identity of a TTR variant. Symptoms consistent with HFpEF, along with concurrent peripheral/autonomic neuropathy, warrant consideration of ATTR as a diagnosis. A complete family history is of great value.
Biopsy
All types of amyloidosis are diagnosed definitively on the basis of demonstration of Congo red binding material in a biopsy or autopsy specimen. Subcutaneous fat aspiration often provides sufficient tissue for diagnosing amyloid, as well as for further studies (eg, immunostaining). Biopsy of an organ with impaired function (eg, heart, GI tract) can definitively establish a cause-and-effect relationship between organ dysfunction and amyloid deposition. See the image below.
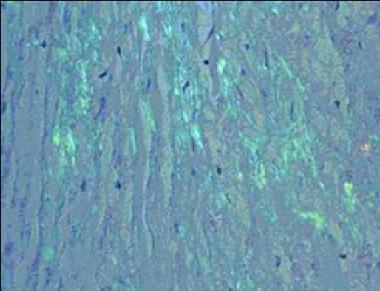 Congo Red staining of a cardiac biopsy specimen containing amyloid, viewed under polarized light.
Congo Red staining of a cardiac biopsy specimen containing amyloid, viewed under polarized light.
Laboratory results for different types of amyloidosis are generally nonspecific, including the following:
Other tests include electrocardiography, nerve conduction studies, and genetic studies (eg, polymerase chain reaction, electrospray ionization mass spectrometry, single-strand conformation polymorphism analysis and/or direct sequencing).
Imaging studies
See Workup for more detail.
No pharmacologic therapy is available that reverses the process of TTR amyloid formation, although studies are ongoing and prior studies have identified drugs that may slow progression of ATTR deposition. There are no FDA approved pharmacological options at this time. Liver transplant remains the gold standard.
Diuretics are the mainstay of therapy for amyloid-related CHF, but must be used with caution due to the restrictive physiology involved. Digoxin and calcium channel blockers are contraindicated in cardiac amyloidosis.
Surgery
Depending on the organ and/or tissue involvement, surgical intervention for patients with ATTR may involve the following:
See Treatment for more detail.
NextThe amyloidoses are a wide range of diseases of secondary protein structure, in which a normally soluble protein forms insoluble extracellular fibril deposits, causing organ dysfunction. All types of amyloid contain a major fibril protein that defines the type of amyloid, plus minor components. Over 20 different fibril proteins have been described in human amyloidosis, each with a different clinical picture (see Amyloidosis, Overview). One such protein that forms human amyloid fibrils is transthyretin (TTR).
TTR acts as a transport protein for thyroxine in plasma. TRR also transports retinol (vitamin A) through its association with the retinol-binding protein. It circulates as a tetramer of four identical subunits of 127 amino acids each. TTR was once called prealbumin because it migrates anodally to albumin on serum protein electrophoresis, but this name was misleading, as TTR is not a precursor of albumin. The TTR monomer contains eight antiparallel beta pleated sheet domains.
TTR can be found in plasma and in cerebrospinal fluid and is synthesized primarily by the liver and the choroid plexus of the brain and, to a lesser degree, by the retina. Its gene is located on the long arm of chromosome 18 and contains 4 exons and 3 introns.[5]
The systemic amyloidoses are designated by a capital A (for amyloid) followed by the abbreviation for the chemical identity of the fibril protein. Thus, TTR amyloidosis is abbreviated ATTR.
In contrast to variant ATTR, normal-sequence cardiac ATTR is associated with aging, usually developing in the seventh and eighth decades of life. This disorder is commonly of little or no clinical significance and only noted on autopsies in studies aiming at estimating its prevalance in an otherwise asymptomatic, aging population. In one autopsy study of people >85 years of age, ATTR was present in 25%.[6] The fraction of autopsied patients with clinically significant symptoms is not known.
The stimuli that lead to normal-sequence ATTR are not understood. Normal-sequence TTR forms cardiac amyloidosis predominantly in men above 60 years of age, a disorder termed senile cardiac amyloidosis (SCA). When it was recognized that SCA is often accompanied by microscopic deposits in many other organs, the alternative name senile systemic amyloidosis (SSA) was proposed. Both terms are now used.[5] The clinical manifestations of severe SCA are similar to those observed in familial ATTR and in cardiac amyloidosis of the immunoglobulin light chain type (AL).
TTR mutations accelerate the process of TTR amyloid formation and are the most important risk factor for the development of clinically significant ATTR. More than 100 amyloidogenic TTR variants cause systemic familial amyloidosis. The age at symptom onset, pattern of organ involvement, and disease course vary, but most mutations are associated with cardiac and/or nerve involvement. The gastrointestinal tract, vitreous, lungs, and carpal ligament are also frequently affected.[5]
ATTR is caused by a single point mutation, of which more than 100 have been described, that promotes destabilazation of the native quarternary structure into B-pleated sheet predominant, insoluble and inactive form. This conformational change hypothesis has been researched in vitro with a key finding that tetramer dissociation is a required and generally rate-limiting step in amyloid fibril formation.
Energetic studies have suggested that amyloidogenic mutations destabilize the native quaternary and tertiary structures of TTR, thereby inducing conformational changes that lead to dissociation of the tetramers into partially unfolded species, which can subsequently self-assemble into amyloid fibrils. However, the wild-type (wt) TTR form can also result in amyloid deposits found in peripheral nerves and cardiac tissue in patients affected by the disease, usually in older patients. It is expected that the process of amyloid aggregation will be further elucidated in the future to address this and other concerns.[7]
When the peripheral nerves are prominently affected, the disease is termed familial amyloidotic polyneuropathy (FAP). When the heart is involved heavily but the nerves are not, the disease is called familial amyloid cardiomyopathy (FAC).
The most common amyloidosis-associated TTR variants in the United States are as follows:
Cardiac ATTR amyloidosis has a progressive increase in prevalence in people older than 80 years and is seen in about 15% of autopsies, with one study finding a prevalence of about 25%. In this setting, the deposited TTR is usually of normal sequence (wt-ATTR).
A few amyloidosis-associated TTR variants are common in certain populations, although few data indicate population frequencies. The most common TTR variants include the following:
Most variants that cause familial ATTR are rare, but a few are common in certain populations. TTR variants are written, according to convention, by the normal amino acid found at a position in the mature protein, followed by the number of the amino acid from the amino terminal end, and the variant amino acid found, using either the three-letter or single-letter amino acid code. The most widely recognized TTR variants are as follows:
Currently, about 100 TTR variants are known, with varying geographic distributions, degrees of amyloidogenicity, and organ predisposition. Currently known TTR variants are listed in the table below.[5] For organ involvement, the following abbreviations are used: PN = peripheral nerves, AN = autonomic nervous system, H = heart, L = liver, LM = leptomeninges, K = kidney, S = skin, E = eye, GI = gastrointestinal tract, CL = carpal ligament, and CNS = central nervous system.
Known TTR Variants (adapted from Connors et al) (Open Table in a new window)
Variant Geographic Focus (Ethnic Origin) Organs Involved Gly6Ser Caucasian None Cys10Arg United States (Hungarian) H, PN, AN, E Leu12Pro United Kingdom CNS, AN, L, LM Asp18Gly United States (Hungarian) CNS, LM Met13Ile Germany None Asp18Asn United States H Asp18Glu South America AN, PN Val20Ile United States, Germany H, CL Ser23Asn United States (Portuguese) H, E, PN Pro24Ser United States PN, H, CL Ala25Ser United States H, PN Ala25Thr Japan CNS, PN Val28Met Portugal AN, PN Val30Met Argentina, Brazil, China, Finland, France, Germany, Greece, Italy, Japan, Portugal, Sweden, Turkey, United States PN, AN, E, LM Val30Ala United States (German) AN, H Val30Leu Japan, United States PN, AN, H, K Val30Gly United States E, CNS, LM Phe33Cys United States CL, E, K, H Phe33Ile Israel (Polish, Ashkenazi Jewish) PN, E Phe33Leu United States (Polish, Lithuanian) PN, AN Arg34Thr Italy PN, H Lys35Asn France PN, H, AN Ala36Pro Greece, Italy, United States (Jewish) PN, E, CNS, CL Asp38Ala Japan H, PN, AN Trp41Leu United States (Russian) E Glu42Gly Japan, Russia, United States PN, AN Glu42Asp France H Phe44Ser United States, Japan PN, H, AN, E Ala45Thr Italy, Ireland, United States H Ala45Asp United States , Ireland, Italy PN, H Ala45Ser Sweden H Gly47Ala Italy, Germany, France PN, H, AN Gly47Arg Japan PN, AN Gly47Val Sri Lanka H, AN, PN, CL Gly47Glu Germany, Italy H, K, PN Thr49Ala France, Italy (Sicily) PN, CL, H Thr49Ile Japan PN, H Thr49Pro United States H Ser50Arg Japan, France, Italy PN, H, AN Ser50Ile Japan PN, H, AN Glu51Gly United States H Ser52Pro United Kingdom PN, AN, H, K Gly53Glu Basque CNS, LM, PN Glu54Gly United Kingdom PN, E, AN Glu54Lys Japan PN, AN, H Leu55Pro United States (Dutch, German), Taiwan PN, E, H, AN Leu55Arg Germany PN, LM Leu55Gln United States (Spanish) AN, E, PN Leu58His United States, Germany H, CL His56Arg United States H Leu58Arg Japan AN, E, CL, H Thr59Lys Italy, United States (Chinese) H, PN, AN Thr60Ala Ireland, United States, Australia, Germany, United Kingdom, Japan H, PN, GI, CL Glu61Lys Japan PN Phe64Leu Italy, United States PN, H, CL Phe64Ser Canada (Italian), United Kingdom CNS, PN, E, LM Ile68Leu Germany, United States H Tyr69His United States, Scotland E Tyr69Ile Japan CL, H Lys70Asn United States, Germany CL, E, PN Val71Ala France, Spain PN, E , CL Ile73Val Bangladesh PN, AN Asp74His Germany None Ser77Tyr Germany, France, United Kingdom PN, H, K Ser77Phe France PN, AN Tyr78Phe France (Italian) PN, CL, S Ala81Thr United States H Ile84Ser United States (Swiss), Hungary H, CL, E, LM Ile84Asn Italy, United States E, H, CL Ile84Thr Germany, United Kingdom PN, AN, H Glu89Gln Sicily PN, H, CL Glu89Lys United States PN, H, AN His90Asn Portugal, Germany None Ala91Ser France PN, H, CL, AN Arg104Cys United States None Arg103Ser United States H Pro102Arg Germany None Ala97Ser China, France, Taiwan H,PN Gln92Lys Japan H Ala97Gly Japan PN,H Gly101Ser Japan None Arg104His Japan, United States (Chinese) None Ile107Met Germany H, PN Ile107Val United States(German), Japan PN, H, CL Ala109Val United States None Ala108Ala Portugal None Ala109Thr Portugal None Ala109Ser Japan PN Leu111Met Denmark H, CL Tyr114Cys Holland PN, E, H, LM, AN, CNS Tyr114His Japan CL Tyr116Ser France PN, CL, AN Thr119Met United States, Portugal None Ala120Ser Afro-Caribbean PN, H, AN Val122Ile Africa, United States, Portugal H Val122Ala United States (Alaska), United Kingdom PN, H, E Deletion of 122Val Ecuador, United States PN, CNS, GI, CL, H Pro125Ser Italy NoneFamilial ATTR was traditionally thought of as a group of autosomal dominant diseases, but it is now known that disease expression is more complicated. The most abundant data pertain to TTR V30M; the following observations have been made:
The explanation for the above observations is not well understood. Other genetic and/or environmental variables are thought to be at play. Anticipation, incomplete penetrance, and clinically sporadic cases in kindreds with unaffected allele carriers also have been observed with other TTR variants.[11]
TTR variants occur in all races.
All TTR variants encoded on chromosome 18 are inherited with equal frequency in males and females. For unknown reasons, disease penetrance is greater and age of onset earlier in males than in females. Individual case reports and several small series suggest that normal-sequence cardiac ATTR is significantly more common in males than in females, although the sex ratio is unknown.[12]
The age of onset varies widely, depending on the presence and identity of the TTR variant.
Morbidity and mortality from ATTR depends on whether a TTR variant is present and, if so, which variant. Some variants cause clinical disease by age 40 years in all gene carriers and are always fatal within a few years of symptom onset. Other variants typically cause much milder, later onset disease, and some carriers of the variant genes remain asymptomatic until late in life.[13]
Morbidity depends on the organ(s) involved. Neuropathy and cardiomyopathy are most common. The most common immediate cause of death is cardiac failure or fatal arrhythmia.[14]
TTR-FAP usually proves fatal within 7–12 years from the onset of symptoms, most often due to cardiac dysfunction, infection, or cachexia.[15]
Within most of the regions in which it is endemic, clinical onset of TTR-FAP often occurs before age 40 years with progressive sensory-motor and autonomic neuropathy, leading to cachexia and eventually death. Length-dependent small-fiber sensory and motor polyneuropathy with life-threatening autonomic dysfunction is a distinguishing feature of TTR-FAP in these areas. In addition, cardiac, renal, and ocular involvement are also common.[16]
In nonendemic areas, and in endemic regions of Sweden, the onset of disease-related symptoms tends to be later in life, from age 50 years onward and with a male predominance for the late-onset TTR-FAP. Neuropathy tends to affect all fibers and may closely resemble chronic inflammatory demyelinating polyneuropathy (CIDP). Typically, sensory and motor neuropathy symptoms of upper and lower extremities occur, associated with mild autonomic symptoms.[16]
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved