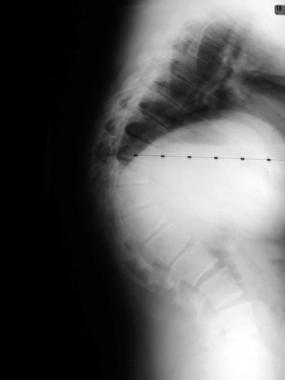
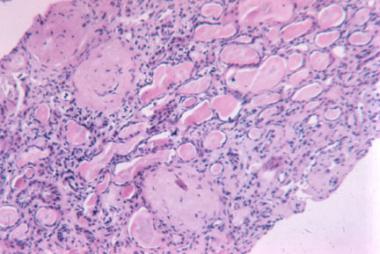
Osteopetrosis is a clinical syndrome characterized by the failure of osteoclasts to resorb bone. As a consequence, bone modeling and remodeling are impaired. The defect in bone turnover characteristically results in skeletal fragility despite increased bone mass, and it may also cause hematopoietic insufficiency, disturbed tooth eruption, nerve entrapment syndromes, and growth impairment. (See Etiology and Presentation.)
Although human osteopetrosis is a heterogeneous disorder encompassing different molecular lesions and a range of clinical features, all forms share a single pathogenic nexus in the osteoclast.[1] Osteopetrosis was first described in 1904, by German radiologist Albers-Schönberg. (See Etiology.)[2]
In humans, 3 distinct clinical forms of the disease—infantile, intermediate, and adult onset—are identified based on age and clinical features. These variants, which are diagnosed in infancy, childhood, or adulthood, respectively, account for most cases. (See Table 1, below.)
Table 1. Clinical Classification of Human Osteopetrosis (Open Table in a new window)
Characteristic Adult onset Infantile Intermediate Inheritance Autosomal dominant[3] Autosomal recessive Autosomal recessive Bone marrow failure None Severe None Prognosis Good Poor Poor Diagnosis Often diagnosed incidentally Usually diagnosed before age 1y Not applicableThe classification of osteopetrosis shown above is purely clinical and must be supplemented by the molecular insights gained from animal models (see Table 2, in Etiology).
Other, rare forms of osteopetrosis have been described (eg, lethal, transient, postinfectious, acquired). A distinct form of osteopetrosis occurs in association with renal tubular acidosis and cerebral calcification due to carbonic anhydrase isoenzyme II deficiency. (See Etiology.)
Overall incidence of osteopetrosis is estimated to be 1 case per 100,000-500,000 population.[1, 4] However, the actual incidence is unknown, because epidemiologic studies have not been conducted.
In infantile osteopetrosis, bone marrow failure may occur. If untreated, infantile osteopetrosis usually results in death by the first decade of life due to severe anemia, bleeding, or infections. Patients with this condition fail to thrive, have growth retardation, and suffer increased morbidity. The prognosis of some patients with infantile osteopetrosis can markedly change after bone marrow transplantation (BMT). Patients with adult osteopetrosis have good long-term survival rates. (See Treatment and Medication.)
Counsel patients with osteopetrosis on appropriate lifestyle modifications to prevent fractures. Provide genetic counseling to patients to allow appropriate family planning. (See Treatment.)
NextTo understand the etiology of osteopetrosis, understanding the bone-remodeling cycle and the cell biology of osteoclasts is essential.
In 1999, Baron clearly and concisely reviewed the cell biology of the bone remodeling.[5] Osteoblasts synthesize bone matrix, which are composed predominantly of type I collagen and are found at the bone-forming surface. Osteoblasts are of fibroblastic origin. Extracellular matrix surrounds some osteoblasts, which become osteocytes. They are believed to play a critical role in the mechanotransduction of strain in bone remodeling.
In contrast, osteoclasts are derived from the monocyte/macrophage lineage. Osteoclasts can tightly attach to the bone matrix by integrin receptors[6] to form a sealing zone, within which is a sequestered, acidified compartment. Acidification promotes solubilization of the bone mineral in the sealing zone, and various proteases, notably cathepsin K, catalyze degradation of the matrix proteins.
Bone modeling and remodeling differ in that modeling implies a change in the shape of the overall bone and is prominent during childhood and adolescence. Modeling is the process by which the marrow cavity expands as the bone grows in diameter. Failure of modeling is the basis of hematopoietic failure in osteopetrosis. Remodeling, in contrast, involves the degradation of bone tissue from a preexisting bony structure and replacement of the degraded bone by newly synthesized bone. Failure of remodeling is the basis of the persistence of woven bone.
For precursor cells to mature, functional osteoclasts require the action of 2 distinct signals. The first is monocyte-macrophage–colony-stimulating factor (M-CSF), which is mediated by a specific membrane receptor and its signaling cascade. The second is the receptor activating NF-kappa B ligand (RANKL), acting through its cognate receptor, RANK. A soluble decoy receptor, osteoprotegerin, can bind RANKL, limiting its ability to stimulate osteoclastogenesis. In mouse models, disruption of these signaling pathways leads to an osteopetrotic phenotype.[7, 8, 9, 10]
The primary underlying defect in all types of osteopetrosis is failure of the osteoclasts to reabsorb bone. A number of heterogeneous molecular or genetic defects can result in impaired osteoclastic function. The exact molecular defects or sites of these mutations largely are unknown. The defect may lie in the osteoclast lineage itself or in the mesenchymal cells that form and maintain the microenvironment required for proper osteoclast function.
The following is a review of some of the evidence suggesting disease etiology and heterogeneity of these causes:
Research has demonstrated that the clinical syndrome of adult type I osteopetrosis is not true osteopetrosis, with the increased bone mass of this condition being due to activating mutations of LRP5.[11] These mutations cause increased bone mass but no associated defect of osteoclast function. Instead, some have hypothesized that the set point of bone responsiveness to mechanical loading is altered, resulting in an altered balance between bone resorption and deposition in response to weight bearing and muscle contraction.
Some cases of type II osteopetrosis result from mutations of CLCN7, the type 7 chloride channel.[12, 13, 14] However, in other families with the clinical syndrome of type II adult osteopetrosis, linkage to other distinct genomic regions has been demonstrated. Therefore, the clinical syndrome is genetically heterogeneous.
In mice, many mutations result in osteopetrotic phenotypes (summarized in Table 2, below). Human homologs are known for only some of the murine lesions.
Table 2. Molecular Lesions Leading to Osteopetrosis in the Mouse (Open Table in a new window)
Gene Protein Lesion Phenotype Human Equivalent Key References Csf1 M-CSF Naturally occurring op allele (frame shift) Reduced size, short limbs, domed skull, absence of teeth, poor hearing, poor fertility, extramedullary hematopoiesis, rescued by administration of M-CSF None known Yoshida et al, 1990 Csf1r M-CSF receptor Targeted disruption in exon 3 Reduced size, short limbs, domed skull, absence of teeth, poor fertility, extramedullary hematopoiesis, slightly more severe than Csf1opphenotype None known Dai et al, 2002 Tnfsf11 RANKL Targeted disruptions Osteopetrosis, failure of lymph nodes to develop None known Kong et al, 1999; Kim et al, 2000 Tnfrsf11a RANK Targeted disruptions Osteopetrosis, failure of lymph nodes to develop Duplications in exon 1 found in Paget disease and in familial expansile osteolysis Li et al, 2000 Ostm1 Osteopetrosis-associated transmembrane protein 1 Naturally occurring deletion Abnormal coat color, short lifespan, chondrodysplasia, failure of tooth eruption, osteopetrosis Infantile malignant osteopetrosis Chalhoub et al, 2003 Acp5 Tartrate resistant acid phosphatase (acid phosphatase 5) Targeted disruption Chondrodysplasia, osteopetrosis None known Hayman et al, 1996 Car2 Carbonic anhydrase II N -ethyl-N -nitrosourea (ENU) mutagenesis No skeletal phenotype in mouse, renal tubular acidosis, growth retardation Osteopetrosis with renal tubular acidosis Lewis et al, 1988 Clcn7 Chloride channel 7 Targeted disruptions Chondrodysplasia, osteopetrosis, failure of tooth eruption, optic atrophy, retinal degeneration, premature death Autosomal dominant type 2 osteopetrosis, autosomal recessive osteopetrosis Kornak et al, 2001; Cleiren et al, 2001 Ctsk Cathepsin K Targeted disruption Osteopetrosis with increased osteoclast surface Pycnodysostosis Saftig et al, 1998; Kiviranta et al, 2005 Gab2 Grb2 -associated binder 2 Targeted disruption Osteopetrosis, defective osteoclast maturation None known Wada et al, 2005 Mitf Micro-ophthalmia–associated transcription factor Spontaneous mutations, ENU mutagenesis, radiation mutagenesis, targeted disruption, untargeted insertional mutagenesis Pigmentation failure, failure of tooth eruption, osteopetrosis, microphthalmia, infertility in both sexes Waardenburg syndrome, type 2a; Tietz syndrome, ocular albinism with sensorineural deafness Hodgkinson et al, 1993; Steingrimsson et al, 1994 Src c-SRC Targeted disruption Osteopetrosis, failure of tooth eruption, premature death, reduced body size, female infertility, poor nursing None known Soriano et al, 1991 Tcirg1 116-kD subunit of vacuolar proton pump Spontaneous deletion, targeted disruption Osteopetrosis, failure of tooth eruption, chondrodysplasia, small size, premature death Autosomal recessive osteopetrosis Li et al, 1999; Scimeca et al, 2000; Frattini et al, 2000 Traf6 Tumor necrosis factor (TNF)-receptor–associated factor 6 Targeted disruptions Osteopetrosis, failure of tooth eruption, decreased body size, premature death, impaired maturation of dendritic cells None known Naito et al, 1999; Lomaga et al, 1999; Kobayashi et al, 2003A distinct form of osteopetrosis occurs in association with renal tubular acidosis and cerebral calcification due to carbonic anhydrase isoenzyme II deficiency. This enzyme catalyzes the formation of carbonic acid from water and carbon dioxide. Carbonic acid dissociates spontaneously to release protons, which are essential for creating an acidic environment required for dissolution of bone mineral in the resorption lacunae. Lack of this enzyme results in impaired bone resorption. Clinical features vary considerably among individuals who are affected.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved