Current achievements in the field of soft tissue tumors are the result of advances in molecular biology, oncogenetics, imaging techniques, immunochemistry, diagnosis by fine-needle aspiration (FNA), surgical reconstruction, radiation therapy, and tissue banking.
The image below depicts needle biopsy of a soft tissue sarcoma.
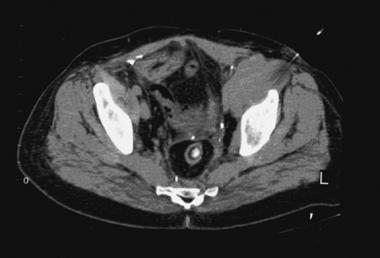 CT-guided needle biopsy of high-grade soft tissue sarcoma arising in left hemipelvis. CT artifact from needle can be seen in upper right corner of image as needle enters lesion just anterior and medial to dome of left hip joint. Image courtesy of Howard A Chansky, MD.
CT-guided needle biopsy of high-grade soft tissue sarcoma arising in left hemipelvis. CT artifact from needle can be seen in upper right corner of image as needle enters lesion just anterior and medial to dome of left hip joint. Image courtesy of Howard A Chansky, MD.
See Soft-Tissue Sarcomas: What You Need to Know, a Critical Images slideshow, to help identify and treat some of these malignant tumors of mesenchymal origin.
Benign soft tissue tumors are fairly common and are treated with surgery alone. Prior to the 1970s, surgery was the primary therapy for malignant soft tissue tumors, and most patients with high-grade tumors had a poor prognosis and a significant mortality. Since the mid-1970s, radiation therapy, chemotherapy, and advanced surgical techniques have helped increase long-term survival and decrease the need for ablative surgery.[1]
Future advances in molecular oncology may further improve diagnostic, prognostic, and treatment protocols for patients with soft tissue sarcomas.[2, 3]
NextSoft tissue is defined as the supportive tissue of various organs and the nonepithelial, extraskeletal structures exclusive of lymphohematopoietic tissues. It includes fibrous connective tissue, adipose tissue, skeletal muscle, blood/lymph vessels, and the peripheral nervous system. Embryologically, most of it is derived from mesoderm, with a neuroectodermal contribution in the case of peripheral nerves.
Soft tissue tumors constitute a large and heterogeneous group of neoplasms. Traditionally, tumors have been classified according to histogenetic features. (Fibrosarcoma, for example, is categorized as a tumor arising from fibroblasts.) However, histomorphologic, immunohistochemical, and experimental data suggest that most, if not all, sarcomas arise from primitive, multipotential mesenchymal cells, which in the course of neoplastic transformation differentiate along one or more lines.
Thus, a liposarcoma appears to arise from a lipoblast but may actually develop through lipoblastic differentiation of a precursor multipotent mesenchymal cell. At the clinical level, soft tissue tumors are classified according to various parameters, including location, growth pattern, likelihood of recurrence, presence and distribution of metastases, patient age, and prognosis.
Although most soft tissue tumors of various histogenetic types are classified as either benign or malignant, many are of an intermediate nature, which typically implies aggressive local behavior with a low-to-moderate propensity for metastasis.
In general, benign soft tissue tumors occur at least 10 times more frequently than malignant ones, though the true incidence of soft tissue tumors is not well documented. However, some insight regarding the incidence of soft tissue sarcomas can be derived from the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) Program, which, between 1973 and 1983, accumulated data on 6883 such tumors.
Overall, the age-adjusted annual incidence of soft tissue sarcomas ranges from 15 to 35 per 1 million population. The incidence increases steadily with age and is slightly higher in men than in women. Malignant soft tissue tumors occur twice as often as primary bone sarcomas.
Approximately 45% of sarcomas occur in the lower extremities, 15% in the upper extremities, 10% in the head-and-neck region, 15% in the retroperitoneum, and the remaining 15% in the abdominal and chest wall. Visceral sarcomas, arising from the connective tissue stroma in parenchymal organs, are not common.
The different types of soft tissue tumors have distinct age distributions. Rhabdomyosarcoma is seen more frequently in children and young adults. Synovial sarcoma arises in young adults. Malignant fibrous histiocytoma and liposarcoma generally occur in older adults. Benign deep masses in adults usually are due to intramuscular lipoma.
In general, the prognosis in older patients with a diagnosis of high-grade sarcoma is poor.
Good evidence exists suggesting that certain genetic disorders and gene mutations are predisposing factors for some benign and malignant soft tissue tumors. The NF1 gene in neurofibromatosis is a classic example, predisposing patients to multiple neurofibromas with a proclivity for malignant transformation. Many tumor suppressor genes, oncogenes, and cytogenetic defects are now associated with various soft tissue sarcomas. Other clinical risk factors account for a small proportion of soft tissue malignancies.
Various cytogenetic abnormalities (see Table 1 below) have been reported to play a significant role in diagnosis, and in the future, some of these abnormalities may become therapeutically significant.
Table 1. Selected Characteristic Cytogenetic Aberrations in Soft Tissue Tumors (Open Table in a new window)
Benign Soft Tissue Tumors CharacteristicSpecific translocations involving selected genes have been observed. One of these, the t(X;18) translocation in synovial sarcoma, results in fusion of the SYT gene from chromosome 18 to either of two highly homologous genes at Xp11, SSX1 or SSX2.SYT-SSX fusion transcript may be detected by reverse transcriptase-polymerase chain reaction (RT-PCR) assay, using a cytologic specimen from FNA biopsy (FNAB), histologic material from paraffin block, or frozen material.
Similar to postirradiation bone tumors, postirradiation fibrosarcomas have been described. The pathogenetic mechanism is the emergence of radiation-induced genetic mutations that encourage neoplastic transformation.
As observed in patients with late-stage breast carcinoma, chronic lymphedema may predispose individuals to the development of lymphangiosarcoma.
An association between exposure to various carcinogens and an increased incidence of soft tissue tumors has been reported. The occurrence of hepatic angiosarcoma, for example, has been linked to arsenic, thorium dioxide, and vinyl chloride exposure.
A classic example of an infection-induced soft tissue tumor is Kaposi sarcoma resulting from human herpesvirus type 8 in patients with human immunodeficiency virus (HIV). Infection with Epstein-Barr virus in an immunocompromised host also increases the likelihood of soft tissue tumor development.
The relation between trauma and soft tissue tumors appears to be coincidental. Trauma probably draws medical attention to a preexisting lesion.
Generally, soft tissue tumors grow centripetally, though some benign tumors (eg, fibrous lesions) may grow longitudinally along tissue planes. Most soft tissue tumors respect fascial boundaries, remaining confined to the compartment of origin until the later stages of development.
Once the tumor reaches the anatomic limits of the compartment, the tumor is more likely to breach compartmental boundaries. Major neurovascular structures usually are displaced as opposed to being enveloped or invaded by tumor. Tumors arising in extracompartmental locations, such as the popliteal fossa, may expand more quickly because of a lack of fascial boundaries; they are also more likely to involve neurovascular structures.
The peripheral portion of the tumor compresses surrounding, normal soft tissue because of centripetal expansile growth. This results in the formation of a relatively well-defined zone of compressed fibrous tissue potentially containing scattered tumor cells. This zone may also consist of inflammatory cells and demonstrate neovascularity.
A thin layer of tissue called the reactive zone surrounds the compression zone, especially in higher-grade tumors. Together, the compression and reactive zones form a pseudocapsule that encloses the tumor and is useful in defining the extent of surgical resection.
Some extremely aggressive lesions with infiltrative growth patterns, such as childhood rhabdomyosarcoma, may not respect anatomic compartmental boundaries and frequently will invade fascial planes.
Soft tissue sarcomas have a propensity for local recurrence. Because recurrences are more difficult to treat than the primary lesion is, complete resection and appropriate use of radiation therapy are critical during the initial treatment. The pseudocapsule provides surgeons with a more or less obvious plane of dissection; however, such an excision can leave behind microscopic or occasionally gross tumor. This may lead to local recurrences in as many as 80% of patients.[4] The addition of postoperative radiation therapy decreases the risk of recurrence associated with a marginal resection.
Technical ease of resectability (and, thus, the likelihood of local control) may be affected by the location of a soft tissue sarcoma. For example, lesions of the head and neck are more likely to involve or abut vital structures; consequently, they often are more difficult to resect than are lesions of the extremities. Even in an extremity, the tumor site may have prognostic implications. For proximal tumors, local control is more difficult to achieve than in tumors located more distally. Retroperitoneal sarcomas, which typically have a poor prognosis, have a higher proclivity for local recurrence and for intra-abdominal dissemination.
The pattern of recurrence generally is predictable, and most tumors destined to recur do so within the first 2-3 years. Adjuvant radiation therapy clearly minimizes local recurrence, but its ability to increase overall chances of survival, though likely, is not certain. Adjuvant chemotherapy may decrease the risk of local recurrence of high-grade tumors, presumably because of a reduction in the size of the tumor and an increase in the reactive zone, but this notion is very controversial.
Regional lymph node involvement is rare in soft tissue sarcomas; fewer than 4% of cases have nodal metastases at presentation. Lymph node involvement is more frequent in epithelioid sarcoma, rhabdomyosarcoma, synovial sarcoma, and clear cell sarcoma. Carcinoma and melanoma should be included in the differential diagnosis for any mass presenting with lymph node metastases.
Many patients with high-grade soft tissue sarcomas, as well as a few with the low-grade type, progress to metastatic disease, even after adequate local control of the primary tumor has been achieved. The lung is by far the most common site of metastasis, which occurs in up to 52% of patients with high-grade lesions.[5]
Although, at the time of presentation, most patients do not have clinically evident metastases, they may have occult micrometastases that eventually manifest clinically. This would appear to be an impetus for the development of chemotherapeutic methods of systemic disease control. At present, however, this is a controversial area of investigation, and it is uncertain whether systemic chemotherapy can improve long-term survival rates for patients with high-grade sarcomas.
A mass is the most common sign of a soft tissue tumor. It usually is painless and does not cause limb dysfunction. However, depending on the anatomic location of the tumor, it may cause pain or neurologic symptoms by compressing or stretching nerves, by irritating overlying bursae, or by expanding sensitive structures. A rapid rate of increase in the size of a mass should arouse suspicion that the lesion is malignant.
Physical examination can be used to determine the location and size of a mass and to exclude other, more common causes of pain. Whether the mass is deep or subcutaneous, transilluminates (cysts), and adheres to underlying structures also can be gleaned from physical examination. Regional lymph nodes should be examined as well. Neurovascular examination is useful for the detection of either primary or secondary tumor involvement.
Extremity masses larger than 5-7 cm and deeper than subcutaneous tissue favor a diagnosis of a malignant soft tissue tumor. However, as many as 30% of soft tissue sarcomas occur in subcutaneous tissue and exhibit relatively less aggressive behavior.[6]
Workup
Copyright © www.orthopaedics.win Bone Health All Rights Reserved