Amyotrophic lateral sclerosis (ALS) is the most common degenerative disease of the motor neuron system. Although ALS is incurable and fatal, with median survival of 3 years, treatment can extend the length and meaningful quality of life for patients.
In 75-80% of patients, symptoms begin with limb involvement. Initial complaints in patients with lower limb onset are often as follows:
Initial complaints with upper limb onset include the following:
With bulbar onset (20-25%), initial complaints are as follows:
Emotional and special cognitive difficulties in some ALS patients are as follows:
Features of more-advanced disease are as follows:
Progression of bulbar disease leads to the following:
The image below illustrates the 4 regions of the body.
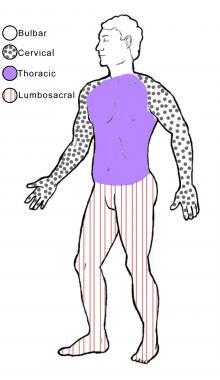 The 4 regions or levels of the body. Bulbar (muscles of the face, mouth, and throat); cervical (muscles of the back of the head and the neck, the shoulders and upper back, and the upper extremities); thoracic (muscles of the chest and abdomen and the middle portion of the spinal muscles); lumbosacral (muscles of the lower back, groin, and lower extremities).
The 4 regions or levels of the body. Bulbar (muscles of the face, mouth, and throat); cervical (muscles of the back of the head and the neck, the shoulders and upper back, and the upper extremities); thoracic (muscles of the chest and abdomen and the middle portion of the spinal muscles); lumbosacral (muscles of the lower back, groin, and lower extremities).
See Clinical Presentation for more detail.
Definitive diagnosis may not be possible with early ALS. Confirmation of the disease may require a period of observation to document its progressive nature and to exclude alternative diagnoses.
The World Federation of Neurology (WFN) has developed a diagnostic algorithm that combines the clinical and, in some cases, electrophysiologic findings.[1] The degree of certainty of diagnosis is increased by the number of body segments that demonstrate upper motor neuron (UMN) and lower motor neuron (LMN) signs. UMN signs are mild weakness, spasticity, and abnormally brisk reflexes; LMN signs are progressive weakness, wasting, and loss of reflexes and muscle tone. WFN categories are as follows:
Hallmark findings in the electrodiagnosis of ALS are normal sensory nerve conduction studies and abnormal motor nerve conduction studies, with reduced motor compound muscle action potentials. The needle exam shows changes characteristic of ongoing denervation and reinnervation of muscles.
In patients with familial ALS, genetic testing may be requested after appropriate counseling. The results of genetic testing may affect not only the patient, but also family members. Tests for the SOD1, TARDBP (coding for TDP-43), FUS, ANG, C9orf72, and FIG4 genes and for the gene causing Kennedy disease are available commercially. Patients with other forms of familial ALS may be referred to centers with a research interest in familial ALS.
See Workup for more detail.
American Academy of Neurology recommendations for management of patients with ALS can be summarized as follows[2, 3] :
Invasive ventilatory support, requiring tracheostomy, may be considered in the following cases:
See Treatment and Medication for more detail.
NextAmyotrophic lateral sclerosis (ALS) is the most common degenerative disease of the motor neuron system. The disorder is named for its underlying pathophysiology, with “amyotrophy” referring to the atrophy of muscle fibers, which are denervated as their corresponding anterior horn cells degenerate. “Lateral sclerosis” refers to the changes seen in the lateral columns of the spinal cord as upper motor neuron (UMN) axons in these areas degenerate and are replaced by fibrous astrocytes (gliosis).
ALS is a fatal disease, with a median survival period of 3 years from onset of weakness.[4] (See Prognosis.) Aspiration pneumonia and medical complications of immobility contribute to morbidity in patients with the disease.
ALS was first described in 1869 by the French neurologist Jean-Martin Charcot and hence is also known as Charcot disease; however, it gained popular recognition and its best-known eponym in the United States after the baseball player Lou Gehrig announced his diagnosis with the disease in 1939.[5, 6, 7, 8, 9] ALS is also known as motor neuron disease (MND).
The cause of ALS is unknown, although a family history of the disease is obtained in about 5% of patients, and twin studies show a genetic contribution with heritability of about 61%.[10] In some cases, ALS overlaps clinically, pathologically, and biologically with frontotemporal dementia, and it may share common biologic mechanisms with Alzheimer disease, Parkinson disease, and other neurodegenerative diseases.[11, 12, 13, 14] (See Etiology.)
ALS is one of the system degeneration diseases, disorders that cause networks that work together in health to disintegrate together in an organized manner.[15, 16] ALS results from the systematic dismantling of the motor neuron system, with the clinical manifestations in each patient deriving from the site of onset and cell type involved; the relative affinity of the dismantling process for prefrontal, upper and lower motor neurons; and the rate of the disease’s spread within the network.[17]
In its classic form, ALS affects motor neurons at 2 or more levels of the motor neuron network supplying multiple regions of the body. It affects lower motor neurons (LMNs) that reside in the anterior horn of the spinal cord and in the brain stem, corticospinal UMNs that reside in the precentral gyrus, and, frequently, prefrontal motor neurons that are involved in planning or orchestrating the work of the upper and lower motor neurons.[18] (See Pathophysiology.)
Loss of LMNs leads to progressive muscle weakness, wasting (atrophy), and fasciculations, with loss of reflexes and muscle tone. Loss of corticospinal UMNs usually leads to milder weakness associated with stiffness (spasticity), which may be severe, and abnormally brisk reflexes.
Loss of prefrontal neurons may result in special forms of cognitive impairment that include, most commonly, executive dysfunction but that may also include an altered awareness of social implications of one’s circumstances and, consequently, maladaptive social behaviors.[19] In its fully expressed forms, the prefrontal dysfunction meets established criteria for frontotemporal dementia.[20, 21] Loss of ability to integrate motor function (apraxia), a premotor function, is seen at times. It is more noticeable in limbs that are not overly weak.
Classic ALS
The term classic ALS is reserved for the form of disease that involves upper and lower motor neurons. The classic form of sporadic ALS usually starts as dysfunction or weakness in one part of the body and spreads gradually within that part and then to the rest of the body.[22] Ventilatory failure results in death 3 years, on average, after the onset of focal weakness. The rate of disease progression varies widely, however, with some patients dying a few months after experiencing their first symptom and others still walking 10 years afterward.
Progressive muscular atrophy and flail limb syndrome
The disease may be restricted to LMNs. When the pattern of LMN involvement is asymmetrical, the disorder is termed progressive muscular atrophy (PMA), and the course is usually indistinguishable from that of classic ALS. Patients with a symmetrical pattern, called flail limb syndrome, have a course that may be far longer.[23]
Primary lateral sclerosis
When only UMNs are involved, the disease is called primary lateral sclerosis (PLS). The course of PLS differs from that of ALS and is usually measured in decades.[24]
Progressive bulbar palsy
Rarely, the disease is restricted to bulbar muscles, in which case it is called progressive bulbar palsy (PBP). In most patients who present with initial involvement of bulbar muscles, the disease evolves to classic ALS.
Familial ALS
Worldwide, a family history of ALS is obtained in about 5% of cases; these patients have familial ALS. Most familial ALS is inherited in an autosomal dominant pattern,[18] often with reduced penetrance, but other patterns, such as X-linked or autosomal recessive inheritance, are seen (see Etiology.)
The fact that in most patients ALS is sporadic does not preclude a genetic contribution to the disease in these cases. ALS as a whole is best thought of as a disease showing complex inheritance.
Complications of ALS can include the following:
The diagnosis of ALS is primarily clinical. Electrodiagnostic testing contributes to the diagnostic accuracy (see Clinical Presentation and Workup). Making a diagnosis is important to patients and families, allowing them to stop the search for alternative causes of a patient's disability and to focus their attention on treatment.
Although ALS is incurable, there are treatments that can extend the length and meaningful quality of life for patients (see Treatment).
Mechanism-specific treatments directed at the processes that cause the disease to evolve after it has expressed itself sufficiently to be diagnosed may, at best, have an ameliorative effect. Treatments that halt the spread of the disease may be more effective than those that try to salvage affected motor neurons. All of these have yet to be realized. Currently, the mainstay of ALS therapy is adaptive treatments directed at the clinical manifestations of the disease.
ALS should not be considered a single disease entity, but rather a clinical diagnosis for different pathophysiologic cascades that share the common consequence of causing preferential progressive loss of motor neurons and the orderly dismantling of the motor neuron system.
Previously, research into the mechanisms resulting in sporadic and familial types of ALS had examined several possibilities. For example, excitotoxicity was suggested to occur secondary to overactivation of glutamate receptors.
Oxidative stress linked to free radical formation was also explored as a cause of ALS, owing to the discovery of mutations in the free radical–scavenging enzyme superoxide dismutase 1 (SOD1).[25] Mitochondrial damage was implicated as a possible mechanism as well, as was autoimmunity to calcium ion channels.
The observation of cytoskeletal proteins in cellular inclusions led to consideration of neurofilament defects as another possible cause of ALS. Inclusions in general implicated defects in the proteasome system were considered as a possible unifying mechanism.
More recent research has focused on RNA processing, because several genetic risk factors for ALS are involved in this metabolic pathway, and aggregation of proteins involved in RNA metabolism has been seen in most forms of ALS. Apoptosis has emerged as a possible method of neuronal death, although this is not certain.
Despite such research, no direct mechanism for ALS has been identified. Most investigators and clinicians agree that various factors, possibly a combination of some or all of the above processes, may lead to development of the disease.[26, 27]
If ALS is considered under the umbrella of neuronal system degenerative diseases, then the specificity for the motor system attacked by the disorder can be attributed to a pathologic process that arises within and spreads through the motor neuron system. Similarly, the focal onset (with subsequent spread) can be compared with the pathogenesis of prion disease (focal onset of a misfolded protein and its spread) or malignancy (a single DNA change or summative mutations, with a final one that confers the pathologic activity and its subsequent spread).
Prionlike propagation of misfolding of proteins—in particular, SOD1 and the 43 kDa transactive response DNA binding protein (TDP-43)—has been proposed as a mechanism for the regional spread of ALS symptoms.[25] The accumulation of misfolded proteins has parallels in other neurodegenerative diseases, including Alzheimer, Parkinson, and Huntington disease.
Motor axons die by Wallerian degeneration in ALS, and large motor neurons are affected to a greater extent than smaller ones. This process occurs as a result of the death of the anterior horn cell body, leading to degeneration of the associated motor axon.
As the axon breaks down, surrounding Schwann cells catabolize the axon's myelin sheath and engulf the axon, breaking it into fragments. This forms small ovoid compartments containing axonal debris and surrounding myelin, termed myelin ovoids. Ovoids then are phagocytized by macrophages recruited into the area to clean up debris.
This type of axonal degeneration can be seen in the brain on biopsy as atrophy and pallor of myelinated motor axons in the corticospinal tracts. In cases in which the disease has been active for a long time, atrophy of the primary motor and premotor cortex may be seen as well. On biopsy of the spinal cord, degeneration of the myelinated motor axons with associated atrophy of the anterior motor roots of the spinal cord can be observed.
Wallerian degeneration also occurs peripherally, and collateral branches of surviving axons in the surrounding area can be seen attempting to reinnervate denervated muscle fibers. On muscle biopsy, various stages of atrophy are noted from this pattern of denervation and subsequent reinnervation of muscle fibers.
In typical ALS, certain motor neurons are spared until very late in the disease process. In the brain stem, these include the oculomotor, trochlear, and abducens nerves. In the spinal cord, the posterior columns, spinocerebellar tracts, nucleus of Onuf (which controls bowel and bladder function), and the Clarke column generally are spared, though the Clarke column can be affected in the familial form of the disease.
Pathways that lead to cell death in ALS may be mediated by the following[28] :
Mutations in the copper/zinc superoxide dismutase 1 (SOD1) gene, which encodes an important antioxidant protein, have been seen in up to 20% of familial ALS patients.[29] Studies in transgenic mice carrying the human SOD1 mutation have provided important information on the pathophysiology of ALS.[28] Also, high levels of oxidative damage to proteins have been found in serum, urine, and cerebrospinal fluid samples from patients, as well as in postmortem samples of patients with both sporadic ALS and SOD1 -familial ALS.[30, 31, 32]
Inferences from animal models, including transgenic models of familial disease, to sporadic human disease are tenuous. However, recognition of the role of glutamate excitotoxicity in sporadic disease and in animal models paved the way to the testing and approval of riluzole, the only treatment that has been shown to ameliorate the course of sporadic ALS, extending patients’ lives by 2-3 months.[33, 34]
The findings below have placed derangements of RNA metabolism at the core of current thinking with regard to the pathophysiology of most types of ALS.
TARDBP gene
In 2006, ubiquitinated inclusions containing pathologic forms of TAR DNA-binding protein-43 (TDP-43) were identified in the cytoplasm of motor neurons of patients with sporadic ALS and in a subset of patients with frontotemporal dementia.[35, 36] TDP-43 is an RNA-processing protein that is normally localized predominantly in the nucleus.
Shortly after their identification in sporadic ALS, TDP-43–positive cytoplasmic inclusions were identified in patients with non-SOD1 familial ALS[37, 38] , and mutations in the gene on chromosome 1 coding for TDP-43 were identified in patients with sporadic and familial ALS.[39, 40, 41, 42, 43, 44]
Mutations in the TARDBP gene, which codes for TDP-43, account for 5% of patients with familial ALS. In addition, TDP-43 inclusions have been found in more than 90% of patients with sporadic ALS, in patients with Guamanian parkinsonism-dementia complex,[45] in the majority of patients with frontotemporal dementia, and in patients with familial British dementia.[46] A review of the continuum of multisystem TDP-43 proteinopathies concluded that the phenotypic expression is linked to the specific cells that are affected by the proteinopathy.[47]
FUS/TLS gene
In 2009, two groups[48, 49] reported that ALS-6, an autosomal dominant form of ALS, results from mutations in the gene for another RNA-processing protein, fused in sarcoma/translated in liposarcoma, or FUS/TLS. (The gene, FUS/TLS, is located on chromosome 16.) Patients these mutations have cytoplasmic inclusions containing FUS/TLS but not TDP-43. Usually, FUS/TLS, like TDP-43, is concentrated in the nucleus. Mutations in FUS/TLS account for 4% of patients with familial ALS.
Additional evidence
Further support for this idea has come from the following[50, 51] :
In 2011, researchers reported that a large hexanucleotide repeat expansion in the noncoding region adjacent to the C9orf72 gene, which is located on the short arm of chromosome 9, accounts for nearly 50% of familial ALS and frontotemporal dementia (FTD) in the Finnish population and more than a third of familial ALS in other groups of European ancestry.[13, 14] It is the most common mutation seen in patients with sporadic ALS. One effect of this mutation is the formation of nuclear RNA foci containing antisense RNA repeats. In addition, a novel mechanism of poly-dipeptide production, repeat-associated non-ATG translation (RAN)[188] has been shown to occur in carriers of the hexanucleotide expansion.[189] The abnormal polypeptides form cytoplasmic deposits. It is unclear how these deposits cause disease. In particular, since median age of onset of C9orf72-associated ALS is the same as that of sporadic ALS, and the deposits precede clinical disease by years, it is unclear how the nuclear or cytoplasmic deposits cause ALS or FTD.
Making the distinction between the pathogenesis and pathophysiology of ALS matters because the mechanisms underlying each of these stages are probably different. This means that interfering with these mechanisms likely requires different approaches. The interventions to prevent disease onset are probably not the same as those required to slow or halt its progression after onset. Prevention of ALS requires modifying or removing factors that are part of disease pathogenesis. A precondition is identifying probable risk factors for ALS.[52]
In contrast to the advances in understanding the pathophysiology of ALS, however, the mechanisms that lead to disease onset (ie, pathogenesis) remain unknown. It is reasonable to assume that the abnormal gene or gene product plays a role in triggering disease onset in familial ALS and may have a role in disease propagation, but having an abnormal gene is neither necessary nor sufficient for developing ALS. Rarely, obligate familial gene carriers do not develop the disease. There is incomplete penetrance, or age-dependent penetrance, of the autosomal dominant genes.
Additional factors must be postulated to intervene between birth and disease onset even in patients who develop familial ALS, because the disease does not appear to start at birth, and within a given family there is great variability in the age of onset. Having normal copies of these genes does not prevent development of sporadic ALS.
Loss of motor neurons bridges disease pathophysiology and its clinical expression. When advanced, this loss results in the characteristic picture seen in cross-sections of the spinal cord in ALS. At the level of the muscle, loss of discrete LMNs causes loss of innervation of individual motor units.
Early in the disease, surviving nerve fibers establish connections and reinnervate motor units that have lost their connection to axons that have died; as a result, larger motor units are formed. These large motor units manifest in histologic stains as fiber-type grouping. (See the image below.) They also have special characteristics on electromyographic testing. Later in the disease, when the motor neurons that supply the large motor units die, group atrophy ensues.
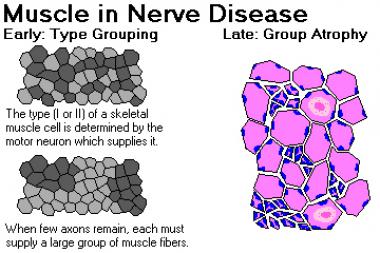 Muscle in nerve disease. Image courtesy of Dr. Friedlander, Associate Professor and Chair of Pathology at Kansas City University of Medicine and Biosciences.
Muscle in nerve disease. Image courtesy of Dr. Friedlander, Associate Professor and Chair of Pathology at Kansas City University of Medicine and Biosciences.
As long as reinnervation can keep up with denervation, clinical weakness may not be detectable, although loss of dexterity may occur. However, as the motor units grow larger and their numbers decrease, the earliest consequence is that affected muscle may fatigue faster than muscle with normal motor units; consequently, one of the first symptoms of ALS may be fatigability of function in the region of onset (for example, “his speech would become muffled toward the end of his sermons”).
As the number of motor units innervating a muscle decreases further, reinnervation can no longer keep up with denervation, permanent weakness develops and progresses, and the affected muscles gradually atrophy. In general, loss of cortical neurons may also result in weakness, but spasticity manifesting as stiffness is a more prominent and disabling UMN symptom.
Acquired nucleic acid changes may trigger disease onset in sporadic ALS.[53] The past 10 years have seen an increase in evidence supporting this hypothesis. This hypothesis relies on the observation that smoking is the only established risk factor for sporadic ALS[54, 55] and provides a mechanism by which smoking might cause the disease—namely, by induction of changes in nucleic acids. It is supported by clinical observation that age-specific incidence of ALS increases with age. It follows a similar logic that suggests that the alkylating components in the cycad are responsible for delayed onset of Western Pacific ALS/PDC.[56, 57]
Simultaneous initial involvement of cortical and spinal motor neurons responsible for the same body part in ALS and the independent spread of disease at spinal and cortical levels has been shown.[58] These observations have been replicated,[59] although other patterns of spread are sometimes seen. They establish an irrefutable role for corticospinal neurons in the early spread of ALS and provide an observational foundation for postulating the existence of a focus of onset and one or more "agents of spread."[60]
Further support for this hypothesis comes from a statistical examination of acquired somatic mutation rates in humans. Somatic mutations inevitably occur during the many cell divisions required for the single-cell zygote to develop into a full organism; those mutations result in genetic mosaicism, and a small focus of genetically altered cells could be a trigger for ALS.[61]
The increase, with age, of age-specific incidence of sporadic ALS suggests that as time passes there is greater chance for changes to accumulate in nucleic acids that will lead ultimately to the development of ALS. This observation has been recently subjected to confirmatory quantitative analysis.[190] This analysis showed that ALS incidence increases with age in a logarithmic fashion, and the slope of log incidence vs. log age is 5. This suggests a multi-step process, analogous to carcinogenesis,{ref 61} with 6 steps needed for ALS to be triggered.
An alternative trigger for disease onset might be appearance of a misfolded intracellular protein that induces other proteins to misfold, with cell-to-cell transmission of the misfolded protein within the motor network. The difference from classic prion disease is lack of transmissibility by inoculation to other organisms, and the confinement to a neuronal system, or network, with a common function. Hereditary or acquired nucleic acid changes may increase the likelihood that a gene product would be susceptible to misfolding, thus triggering disease onset.
The recent identification of the non-coding C9ORF72 hexanucleotide expansion in many familial and some sporadic ALS cases opens up a possibility that failure of regulation of a normal gene product may underlie ALS initiation and spread.[62, 63, 64, 65, 66, 67] In a broader sense, generation of a faulty regulatory gene product responsible for motor network maintenance, or failure to regulate such a product, may account for the specific disintegration of the motor network. MicroRNAs are particularly attractive candidates for this role.
The affirmation of spread substantiates the concept of a biologic focal onset for ALS. This in turn lends credibility to the concept of a focal trigger for ALS onset that generates the production of 1 or more agents of spread.[53]
Under this hypothesis, disease phenotype in each patient depends on the site of onset and the relative affinity of the specific agent of spread in that patient to motor neurons at the different hierarchical levels of the motor system (prefrontal, corticospinal, spinal/bulbar).[60] The concept of preferential affinity of the agent of spread may apply even to specific motor neurons within a given hierarchy, resulting, for example, in the special phenotypes in which LMNs are predominantly affected (flail arm syndrome, flail leg syndrome).[68]
Most of the biochemical changes found in the upper and lower motor neurons of diseased patients are probably downstream from those that initiate the disease and cause its spread. Some of these changes may represent the processes by which the motor neurons die, but others may reflect the efforts of the motor neurons to survive by compensating for, or “fighting,” the primary pathologic processes driving progression of ALS.
Most ALS cases are sporadic, and the specific cause of sporadic ALS is unknown. Many abnormal genes have been identified in familial cases and are considered causal, although the precise mechanism by which they cause ALS is unknown for most.
All of the mutated genes found to give rise to familial ALS have also been found in patients with sporadic ALS. This is to be expected, as the distinction between familial and apparently sporadic disease is based on obtaining a family history, which in turn depends on gene penetrance, family size, age of family members, and the level of knowledge of the person being interviewed.[69]
In addition, first-degree relatives of patients with apparently sporadic disease have an increased risk of ALS. The overall lifetime risk of ALS in these relatives is low, however (approximately 1 in 50).[70]
A family history of ALS is obtained in about 5% of cases and is often consistent with a Mendelian autosomal dominant pattern of inheritance. While most cases of familial ALS are indistinguishable from sporadic disease, others have unique phenotypes.[71]
Juvenile forms of ALS are more commonly familial. Mean age of onset is 10-20 years younger in patients with familial ALS than in patients with apparently sporadic disease, and variability in age of onset between families is greater than variability within families.[18] Age of onset may also be modified by genetic factors independent of the cause of ALS.[72, 73]
Many specific heritable gene mutations have been described in familial ALS (see Table 1, below).[8] A curated, up-to-date list that includes unpublished mutations, genotype-phenotype correlations, and tools for analysis is maintained on the ALSoD website.
Table 1. Familial Forms of ALS[74, 75, 76] (Open Table in a new window)
Gene Locus Protein Inheritance SOD1About 10-20% of familial ALS cases result from a mutation in the copper/zinc superoxide dismutase 1 (SOD1) gene, also known as ALS1. In general, SOD1 ALS is a LMN form of the disease.[77] The other genes most commonly involved in familial ALS are C9orf72, FUS (ALS6) and TARDBP (ALS10).
SOD1 mutations
More than 140 allelic variations have been seen in SOD1. Some of these variations are characterized by a relatively predictable age of onset or rate of disease progression.
The traditionally used numbering of the amino acids in SOD1, which omits the start codon, is no longer in line with standard practice but will be used here. To translate to the modern numbering, an additional codon should be counted. For example, the D90A mutation frequent in Scandinavia should be p.D91A.
The most common SOD1 mutation in the United States is the A4V mutation, accounting for 50% of SOD1 ALS cases. It causes a rapidly progressive lower motor disease with a mean survival period of 1 year. The North American SOD1 A4V mutation descended from 2 founders (Amerindian and European) 400-500 years ago.[78]
Not all individuals with an SOD1 mutation develop ALS. SOD1 ALS has been shown to be a gain-of-function disease; knockout mice depleted of SOD1 do not develop ALS, and transgenic mice with 1 mutated and 2 normal genes have worse disease than those with 1 normal and 1 mutated gene. Misfolding and precipitation of the abnormal (and normal) SOD1 proteins are thought to be part of the pathophysiology of SOD1 ALS, but why the disease begins when it does and how it causes the LMN ALS phenotype is not clear.
Work in animal models suggests that silencing from birth of the expression of mutant SOD1 by silencing RNA molecules (siRNA) may prevent disease onset in transgenic mouse models.[79] This is an exciting direction for research in humans with SOD1 mutation, but major barriers need to be overcome first, including demonstration that this approach is effective in halting the disease after its clinical onset.[71, 74, 75, 80]
TARDBP and FUS mutations
Gene mutations resulting in abnormalities in proteins that regulate RNA processing have been discovered in patients with autosomal dominant familial ALS. Mutations in the TARDBP gene, which codes for TDP-43, have been found in 5% of patients with familial ALS.[37, 38, 39, 40, 41, 42, 43, 44] Mutations in the FUS gene are found in 3-4% of familial ALS cases.[71]
The mechanism by which these mutations cause ALS is distinct from that which takes place in SOD1 mutations. Although ubiquinated pathologic TDP-43 aggregates have been found in motor neuron cytoplasm of patients with sporadic ALS, they are not specific for this disease and have been found in affected nonmotor cells in patients with Guamanian parkinsonism-dementia complex,[45] British familial dementia,[46] and Alzheimer disease,[81] as well as in most patients with frontotemporal dementia.
Thus, formation of pathologic TDP-43 and its ubiquination may prove to be a mechanism of cell death that is not specific to ALS and is triggered by upstream processes, causing clinical pathology that depends on the cells affected. Conversely, TDP-43 deposition may prove to be a nonspecific defense mechanism involving an unsuccessful attempt to mitigate the action of the true instigators of cell death in a spectrum of neurodegenerative diseases, or it may be an epiphenomenon common to many forms of neurodegenerative diseases. Transgenic animal models with mutated TDP-43 do not develop ALS.
C9orf72 mutation
Studies of families with familial ALS in which some individuals also had frontotemporal dementia found linkage to a region on chromosome 9p21[82, 83, 84] Subsequently, a large genome-wide association study (GWAS) identified single-nucleotide polymorphisms (SNPs) associated with apparently sporadic ALS in the same region. This was confirmed in a further GWAS-wide association study of patients with apparently sporadic ALS and controls from 8 countries,[85] as well as in a Finnish study[86] using family-based samples.
In 2011, two groups reported the discovery of a hexanucleotide (GGGGCC) repeat expansion in the first intron of the chromosome 9 open reading frame 72 (C9orf72) gene—the function of which is as yet unknown—as the cause of chromosome 9p21–associated ALS and frontotemporal dementia.[62, 63] This expansion of hexanucleotide repeats (from ≤23 in normal individuals to thousands in affected individuals) appears to be the most common genetic abnormality in ALS and in frontotemporal dementia.
The repeat expansion was found in 46.0% of patients with familial ALS, 21.1% with sporadic ALS, and 29.3% with familial frontotemporal dementia, in the Finnish population.[63] In their analysis of an extended North American clinical series, DeJesus-Hernandez et al found the C9orf72 expansion in 23.5% of patients with ALS cases and 11.7% of patients with familial frontotemporal dementia.[62]
More recent reports have found the C9ORF72 repeat expansion in 22-57% of familial ALS patients (depending in part on geographic origin) and in 3.6-7% of sporadic ALS patients.[64, 65, 66, 67] The frequency of the mutation varies widely even within Europe.[87]
Haplotype analysis of 5 European cohorts has shown that the hexanucleotide repeat expansion in C9orf72 had a single founder and arose around 6300 years ago.[87] The haplotype from which the mutation arose is intrinsically unstable, with an increased number of repeats.[87]
The phenotype of C9orf72 -mediated ALS shows distinct pathology, with p62-positive, TDP43-negative inclusions in the cerebellum and hippocampus.[88] Clinically, patients with this mutation have an earlier onset of disease and are more likely to have bulbar-onset disease, cognitive and behavioral impairment, and a family history of frontotemporal dementia than are ALS patients who carry other known mutations.
Other genes in familial ALS
Mutation in the ubiquilin 2 (UBQLN2) gene has been identified as a cause of X-linked dominant familial ALS and ALS with frontotemporal dementia.[89] This finding is of interest because it directly implicates the proteasome pathway in ALS pathogenesis.
Mutation in the profilin 1 (PFN1) gene has been identified in families with familial ALS.[76] The protein encoded by PFN1 plays a critical role in the conversion of monomeric (G)-actin to filamentous (F)-actin. Thus, the identification of this mutation provides further support for the role of the cytoskeleton and axonal transport in ALS pathogenesis.
The most widely accepted hypothesis regarding the cause of sporadic ALS posits that interactions between genetic, environmental, and age-dependent risk factors trigger disease onset. (See the image below.) ALS shows complex inheritance, which means that Mendelian, non-Mendelian, and apparently sporadic patterns of inheritance are seen. Smoking is the only environmental risk factor identified to date that may be considered “established.”[54, 55]
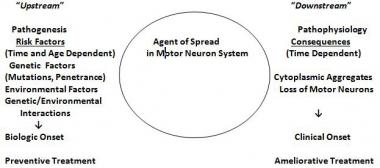 The genetic/environmental/age- and time-dependent interactions hypothesis for amyotrophic lateral sclerosis (ALS). Risk factors operate upstream to a putative biochemical transformation (likely an acquired nucleic acid or protein change), which causes the appearance of altered proteins or nucleic acids or abnormal quantities of normal proteins or nucleid acids. These agents spread within the motor system and cause the downstream disintegration of the motor system and the downstream biochemical, histologic, and clinical consequences of ALS. (Adapted from Armon C. What is ALS? In: Amyotrophic Lateral Sclerosis: A Patient Care Guide for Clinicians. Bedlack RS, Mitsumoto H, Eds. Demos Medical Publishing, New York, 2012:1-23)
The genetic/environmental/age- and time-dependent interactions hypothesis for amyotrophic lateral sclerosis (ALS). Risk factors operate upstream to a putative biochemical transformation (likely an acquired nucleic acid or protein change), which causes the appearance of altered proteins or nucleic acids or abnormal quantities of normal proteins or nucleid acids. These agents spread within the motor system and cause the downstream disintegration of the motor system and the downstream biochemical, histologic, and clinical consequences of ALS. (Adapted from Armon C. What is ALS? In: Amyotrophic Lateral Sclerosis: A Patient Care Guide for Clinicians. Bedlack RS, Mitsumoto H, Eds. Demos Medical Publishing, New York, 2012:1-23)
Several lines of evidence support the hypothesis that genetic risk factors may influence disease initiation in apparently sporadic ALS, apart from finding known Mendelian gene mutations in patients with no family history. Twin studies show a genetic contribution to apparently sporadic ALS, with heritability of 0.61.[10] Increased risk for ALS and (in some studies) clustering of non-ALS neurodegenerative disease is found in relatives of patients with apparently sporadic ALS.[70, 90, 91, 92]
Smoking
Cigarette smoking is the only exogenous risk factor that may be considered an established risk factor for ALS[54] (level A conclusion, based on 3 class II studies[93, 94, 95] and 1 class III study[96] ). In addition, a population-based study from the Netherlands demonstrated that current smokers are at increased risk for ALS, with an odds ratio of 1.38, and have shorter survival.[55]
Some aspects of the findings in these studies suggest that smoking may be implicated directly in causing the disease. Overall, studies have shown that active smokers have approximately double the risk of developing ALS compared with never smokers. Former smokers have an intermediate risk.
Identifying smoking as an established risk factor for ALS has the following major implications:
Focusing on processes at the initiation of sporadic ALS and close to its initiation, in order to account for its early spread within the motor system, may provide new avenues to treatments to stop its progression. This approach may augment the current focus on processes that occur relatively late in the course of the disease and cause the death of motor neurons directly.
No other risk factor has attained the level of certainty that smoking has with regard to its association with ALS. Trauma, physical activity, residence in rural areas, and alcohol consumption are probably not risk factors for ALS.[52] Indeed, in the population-based study from the Netherlands noted above, current alcohol consumption was associated with a reduced risk of ALS.[55]
Military service
Putative risk factors include service in the US military during World War II, the Korean War, and Vietnam,[97] as well as deployment to the Persian Gulf in the 1991 Persian Gulf War.[98] However, close scrutiny has cast doubt on the quality of the evidence supporting the role of these factors in triggering ALS.[98, 99, 100, 101, 102] More recently, a 13-year follow-up study found no excess of ALS among Gulf War veterans.[103]
Other putative exogenous risk factors have not risen to the level of probable. These include exposure to pesticides,[104] postmenopausal hormone use,[52, 105] and physical exercise.[106]
Sports
A possible increased risk of ALS in Italian professional soccer players was reported in 2005.[107, 108] Initially, it appeared that the apparent increase in risk may have resulted from underestimation of the expected number of cases of ALS.[109, 110, 111, 112, 113] However, a 5-year extension of the follow-up of this cohort showed an unambiguous excess of cases of ALS; 8 cases (including 3 new ones) were reported, even though the number of expected cases was 1.24.[114]
However, the ALS in the professional soccer players displayed atypical features; ie, 5 of the 8 cases had a bulbar onset, and 5 had been diagnosed between 2000 and 2006 (compared with 3 between 1980 and 1999 and none between 1970 and 1979). The authors concluded that the predilection for soccer players to develop ALS derives from a complex interplay between genetic predisposition for physical endurance and external factors such as drugs or herbicides.[114]
A subsequent report suggested that a form of motor neuron disease might develop in individuals, such as boxers, who have sustained repetitive injuries to the brain and developed chronic traumatic encephalopathy (CTE).[115] However, the claim that this was a novel form of motor neuron disease was challenged; instead, the possibility that these cases represented coincidental co-occurrence of ALS was proposed as more plausible.[116, 117, 118]
More recently, a study in retired US National Football League (NFL) players showed that, while their overall mortality was 50% less than expected, their mortality from neurodegenerative diseases was higher than expected. In particular, mortality from ALS and Alzheimer disease was 4-fold higher than expected.[119] These results are based on 7 individuals who died with Alzheimer disease and 7 individuals who died with ALS, out of a cohort of 3439 individuals.
The interpretation of this number as excessive appears to be a consequence of the method used to calculate the expected rate,[109] which may result in underestimation of that rate. In addition, when an apparent excess of neurodegenerative disease appears in the context of greatly reduced overall morality, the emergence of neurodegenerative deaths due to loss of competing causes of mortality needs to be considered.
These reports raised the question whether trauma to the head may be a risk factor for ALS. An evidence-based review of the literature concluded that for instances of isolated head trauma this was not the case.[120]
A subsequently published national population–based case-control study from Sweden found no association of ALS with severe head injury occurring more than 3 years before ALS diagnosis, nor was ALS associated with subtypes of head injury or repeated injuries occurring more than 3 years before diagnosis.[121] Exclusion of injuries occurring within 3 years of diagnosis is necessary to have some assurance that the injury occurred before biologic onset of the disease, which likely precedes clinical onset by several years.[122]
A population-based study from Rochester, Minnesota, showed no increased risk of neurodegenerative diseases among 438 players who played American football in high school between 1946 and 1956, despite poorer protective equipment and less regard for concussions compared with today, and no rules prohibiting head-first tackling.[123]
The balance of the evidence supports the conclusion that head trauma in general, including repetitive head trauma, is not a risk factor for ALS. The special circumstances of the Italian soccer players are uncertain, as their form of ALS was unusual, with most experiencing bulbar onset.
The special circumstances of the NFL players are also uncertain, as the data are sparse, and it is not clear that the numbers represent a true excess. Risk factors other than the sport itself may be involved, including local environmental risk factors or ingestion of testosterone, anabolic steroids, or other drugs.[114, 124]
One report indicated that head injury does not alter disease progression or neuropathological outcomes in patients with ALS.[191]
Most of the research on Western Pacific ALS has focused on Guam. Ingestion of food products derived from the false sago palm, Cycas micronesica (recently separated from Cycas circinalis), was proposed by nutritional anthropologist Marjorie Whiting as the process predisposing to the development of this form of ALS.[125] Despite the cycad nut being subjected to an elaborate preparation process to rid it of toxins before being used as a substrate for flour, some toxic factor was presumed to remain.[126]
In addition, the cycad nut is consumed by the flying fox (a type of bat), which used to be part of the Chamorro peoples' diet on Guam. Toxins from the cycad nut may have been concentrated (bioamplified) in the bat and delivered to the human consumer. The consumption of the flying fox was higher in the mid-20th century than it is now.[127] Most flying foxes consumed in Guam currently are imported.
An epidemiologic study in Guam provided evidence consistent with that hypothesis; its conclusions have been challenged,[127] but the challenges themselves have been questioned.[126]
The nature of the putative toxic component of cycad that may be responsible for delayed-onset neurodegenerative disease has also been a matter of intense debate. One hypothesis is that cycad contains excitotoxic amino acids that do not exert an effect until many years after they have been ingested.[128, 129, 130]
An alternative hypothesis is that alkylating components induce changes in nucleic acids[56, 57] that increase the likelihood that subsequent, additional, age-dependent nucleic acid changes trigger disease onset in Guamanian ALS/Parkinson-dementia complex (ALS/PDC).
Approximately 5600 people in the United States are diagnosed with ALS each year. The annual incidence is 2-3 per 100,000 population; this is about equal to that of multiple sclerosis and 5 times higher than that of Huntington disease. It is estimated that as many as 18,000 Americans may have ALS at any given time.
The lifetime risk for developing ALS for individuals aged 18 years has been estimated to be 1 in 350 for men and 1 in 420 for women.[109] These estimates are close to those reported from 4 European registries, using different methods.[131, 132, 133]
Age-adjusted European incidence data are similar to those for members of the US population who are of European descent.[131, 134] Most variability between countries has been attributed to different age composition or differences in case finding. More recent data, however, suggest that ethnic variability in disease incidence exists[135, 136] that may not be explained entirely by differences in case finding, with lower incidence in nonwhites or individuals of mixed ethnicity. Although this possibility is not supported by all studies, it merits further examination.[137]
Finland has one of the highest rates of ALS in the world; the disease occurs in the Finnish population nearly twice as frequently as it does in other populations of European ancestry.[86] A study from Finland found 2 clusters of cases based on geographic location at time of death and a cluster based on time of birth that closely matched one of the time-of-death clusters.[138] It was recognized that these results could be consistent with either a genetic or environmental cause. With the discovery of the hexanucleotide repeat expansion in C9orf72, most cases of ALS in Finland have been confirmed to be due to genetic factors.
In the United States, ALS affects whites more often than nonwhites; the white-to-nonwhite ratio is 1.6:1.[135] Uncertainty surrounds this finding, however, as it has been considered to be an artifact of reduced case-finding in nonwhites. More convincing evidence for racial differences has come from an epidemiologic study in Cuba.[136]
Small population clusters have been identified that have higher rates of ALS. The Chamorro people of Guam and Marianas Island, residents of the Kii peninsula of Japan’s Honshu Island, and the Auyu and Jakai people of southwest New Guinea have a higher incidence of ALS than is found in populations elsewhere in the world.[8] The Chamorro population in Guam in the mid-20th century had an annual incidence of ALS (often in association with parkinsonism and dementia) as high as 70 cases per 100,000 (see Pathophysiology).[128] The incidence has since decreased to 7 cases per 100,000.
For most of the lifespan, the incidence of ALS is higher in men than in women, with an overall male-to-female ratio of 1.5-2:1.[18] Later in life, the incidence tends to equalize; this occurs at age 40-50 years in some populations and after the age of 65-70 years in others.[139]
Onset of ALS may occur from the teenage years to the late 80s; the incidence rises with increasing age until approximately age 75-80 years. Mean age of onset of sporadic ALS is 65 years; mean age of onset of familial ALS ranges from 46-55 years.
ALS is a fatal disease. Median survival is 3 years from clinical onset of weakness. However, longer survival is not rare. About 15% of patients with ALS live 5 years after diagnosis, and about 5% survive for more than 10 years. Long-term survival is associated with a younger age at onset, being male, and limb (rather than bulbar) symptom onset. Rare reports of spontaneous remission exist.[140] In familial ALS that results from an alanine-to-valine mutation in codon 4 of the SOD1 gene (A4V mutation), average survival is 12 months from disease onset.[78]
Regionally limited forms of motor neuron disease (ie, brachial biplegia, lumbosacral biplegia, and progressive bulbar palsy [PBP] that remains restricted[141] ) progress slower than does classic ALS. Progressive muscular atrophy (PMA), distinct from classic ALS because of lack of upper motor neuron (UMN) findings, progresses at the same rate as classic ALS. UMN-predominant ALS progresses at a slower rate. Survival in cases of primary lateral sclerosis (PLS) is measured in decades. These observations suggest that it is the loss of LMNs that determines the prognosis.
Frontotemporal executive dysfunction may precede or follow the onset of ALS, but most patients with ALS do not have overt dementia, and cognitive impairment is usually subtle.[142] Approximately 15% of patients with ALS meet criteria for frontotemporal dementia (FTD). Patients with ALS associated with FTD have shorter survival than do those with ALS alone.[143, 144]
The most effective predictor of survival is the rate of observed disease progression, estimated via the time between symptom onset and diagnosis or evaluated by employing measures such as the ALS Functional Rating Scale (ALSFRS).[145] Estimates derived as a ratio in which the numerator is a measurement of functional loss—as obtained using the ALSFRS-Revised (ALSFRS-R), the forced vital capacity (FVC) as percent of predicted, strength, or motor unit number estimates (MUNEs)—and the denominator is time elapsed from disease onset to the time of measurement, may provide more individualized prognostic information.[146, 147, 148, 149, 150]
Roche et al have proposed a system of stages, the timing of which is standardized as proportions of elapsed time through the course of ALS.[151] The milestones, and their typical time of occurrence, are as follows:
It may be seen that most patients are diagnosed at a point at which ALS has extended to involve at least 2 regions and that the needs for gastrostomy and noninvasive ventilation (NIV) are usually recognized almost at the same time. These percentages are consistent with the figures of 12 months’ mean time from onset to diagnosis and 3 years’ mean duration of disease from onset to death.
Most patients with PMA have a course indistinguishable from that of patients with classic ALS (except for the absence of UMN findings). However, some patients may have a longer course, particularly those with flail arm or flail leg syndrome.[68]
Kim et al concluded that PMA should be considered a form of ALS.[152] Review of the medical records of 91 patients with PMA and 871 with ALS showed that patients with PMA were more likely to be male, to be older, and to live longer than those with ALS, but risk of death increased with age at onset in both patient groups and UMN signs developed in 22% of patients with PMA within 61 months after diagnosis.[152]
In the study, demographic and other clinical variables did not differ at diagnosis between patients who did or did not develop UMN signs. In PMA, as in ALS, the factors present at diagnosis that predicted shorter survival were greater number of body regions affected, lower FVC, and lower ALSFRS-R score.
Patients the author has surveyed have indicated ambivalence about being offered individualized information early in the course of the disease (when it may matter most).[153] The author does not offer patients individualized prognostic information at the first visit. When patients do request it, he asks them to consider the possible implications of the answers they may receive and to ask him again at a subsequent visit. It is also helpful to discuss the implications of the rate of disease progression, based on the joint observations of the patient and the physician.
The following education resources are available to patients with ALS:
Informational web sites include the following:
For patient education information, see the Brain and Nervous System Center, as well as Amyotrophic Lateral Sclerosis (ALS, Lou Gehrig’s Disease) and Advance Directives.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved