Dermatomyositis is an idiopathic inflammatory myopathy with characteristic cutaneous findings that occur in children and adults (see the image below). This systemic disorder most frequently affects the skin and muscles but may also affect the joints; the esophagus; the lungs; and, less commonly, the heart.[1, 2] Dystrophic calcinosis may complicate dermatomyositis and is most often observed in children and adolescents.
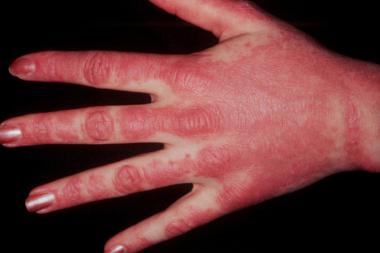 These lesions on dorsal hands demonstrate photodistribution of dermatomyositis. Note sparing of interdigital web spaces.
These lesions on dorsal hands demonstrate photodistribution of dermatomyositis. Note sparing of interdigital web spaces.
Persons with dermatomyositis often present with skin disease as one of the initial manifestations, and it may be the sole manifestation at onset in perhaps as many as 40% of individuals with this condition. Cutaneous involvement may manifest as follows:
Muscle disease may occur concurrently, may precede the skin disease, or may follow the skin disease by weeks to years. Muscle involvement manifests as the following:
Systemic manifestations that may occur include the following:
See Clinical Presentation for more detail.
Examination for cutaneous dermatomyositis may reveal the following findings:
Examination for muscle disease in dermatomyositis may demonstrate the following:
Testing
Laboratory and other studies that may be helpful include the following:
Imaging studies
The following imaging studies may be used in the evaluation of dermatomyositis:
Procedures
The following procedures may be helpful in the evaluation of dermatomyositis:
See Workup for more detail.
Therapy for the muscle component of dermatomyositis involves the use of corticosteroids, typically with an immunosuppressive agent. Therapy for the skin disease includes the following, among other options:
Pharmacotherapy
Medications used in the management of dermatomyositis include the following:
In addition to the medications listed above, diltiazem, colchicine, alendronate, and warfarin are among the medications that have shown potential benefit in treating calcinosis. Surgical excision of focal, tender calcinotic lesions is also considered a therapeutic option.
Nonpharmacotherapy
General therapeutic measures may include the following:
Surgery
Surgical care is usually unnecessary in the management of dermatomyositis. However, some patients may benefit from surgical removal of localized areas of calcinosis, particularly those that are painful.
See Treatment and Medication for more detail.
NextDermatomyositis is an idiopathic inflammatory myopathy (IIM) with characteristic cutaneous findings. It is a systemic disorder that most frequently affects the skin and muscles, but may also affect the joints; the esophagus; the lungs; and, less commonly, the heart.[1, 2] Calcinosis is a complication of dermatomyositis that is observed most often in children and adolescents. An association between dermatomyositis and cancer has long been recognized in adult patients.[5, 6, 7, 8, 9, 10]
In 1975, Bohan and Peter first suggested a set of five criteria to aid in the diagnosis and classification of dermatomyositis.[11, 12] Four of the five criteria are related to the muscle disease, as follows:
The fifth criterion is compatible cutaneous disease.
In addition to dermatomyositis, Bohan and Peter suggested the following four subsets of myositis[12] :
In a subsequent publication, Bohan et al noted that cutaneous disease may precede the development of the myopathy in patients with dermatomyositis.[11] In addition, the existence of another subset of patients with dermatomyositis that affects only the skin (ie, amyopathic dermatomyositis [ADM], or dermatomyositis sine myositis) has been recognized. Finally, another subset of patients with dermatomyositis are those with controlled myopathy who continue to have severe and sometimes debilitating skin disease (ie, postmyopathic dermatomyositis).
ADM is diagnosed in patients with typical cutaneous disease who show no evidence of muscle weakness and in whom serum muscle enzyme levels are repeatedly normal over a 2-year period in the absence of the use of disease-modifying therapies such as corticosteroids, immunosuppressive agents, or both for 2 months or longer.
When studied, some ADM patients may have abnormal findings on ultrasonography, electromyography, magnetic resonance imaging (MRI), magnetic resonance spectroscopy, or muscle biopsy. These patients are better classified as having hypomyopathic dermatomyositis. ADM or hypomyopathic DM may also be related to an underlying malignancy.
The term clinically amyopathic dermatomyositis (CADM) is often used to encompass patients with both amyopathic and hypomyopathic dermatomyositis.[13] CADM is estimated to account for about 20% of patients with dermatomyositis,[14] and one large review suggests that CADM is associated with malignancy and lung disease as frequently as classic dermatomyositis.[15] In addition, some patients with CADM develop severe pulmonary disease, particularly persons from Asian countries.[16]
Patients exist in whom myositis resolves after therapy but skin disease remains an active and important feature of the disorder. These patients are not classified as having ADM, even though by this point, the skin is the major and often only manifestation of the disease. Germani and colleagues have suggested the term postmyopathic dermatomyositis for these patients.[17]
Therapy for the muscle component of dermatomyositis involves the use of systemic corticosteroids, with or without an immunomodulatory agent. The skin disease is treated with sun avoidance, sunscreens, photoprotective clothing, topical corticosteroids, antimalarial agents, methotrexate, mycophenolate mofetil, or intravenous immunoglobulin (IVIG). Systemic corticosteroids are generally not administered for cutaneous involvement. Rituximab may be useful in the treatment of muscle disease in dermatomyositis, and has had mixed results in treatment of skin disease.[18, 19]
Physical therapy and rehabilitative measures are necessary in selected patients. Sun protective measures are necessary for patients with skin disease. Patients may visit The Myositis Association Web site for more information.
The prognosis of dermatomyositis depends on the severity of the myopathy, the presence of malignancy, and/or the presence of esophageal and/or cardiopulmonary involvement. Residual weakness is common, even in patients who fully recover.
For discussion of dermatomyositis in pediatric patients, see Juvenile Dermatomyositis.
Dermatomyositis is considered to be the result of a humoral attack against the muscle capillaries and small arterioles (endothelium of the endomysial blood vessels). Since 1966, there has been evidence supporting an ongoing microangiopathy.[20]
The disease starts when putative antibodies or other factors activate C3, forming C3b and C4b fragments that lead to formation of C3bNEO and membrane attack complex (MAC), which are deposited in the endomysial vasculature. Complement C5b-9 MAC is deposited and is needed in preparing the cell for destruction in antibody-mediated disease. B cells and CD4 (helper) cells are also present in abundance in the inflammatory reaction associated with the blood vessels.
As the disease progresses, the capillaries are destroyed, and the muscles undergo microinfarction. Perifascicular atrophy occurs in the beginning; however, as the disease advances, necrotic and degenerative fibers are present throughout the muscle.
The pathogenesis of the cutaneous component of dermatomyositis is poorly understood, but is thought to be similar to that of muscle involvement.
Studies on the pathogenesis of the muscle component have been controversial. Some suggest that the myopathy in dermatomyositis is pathogenetically different from that in polymyositis. The former is probably caused by complement-mediated (terminal attack complex) vascular inflammation, the latter by the direct cytotoxic effect of CD8+ lymphocytes on muscle. However, other cytokine studies suggest that some of the inflammatory processes may be similar. One report has linked tumor necrosis factor (TNF) abnormalities with dermatomyositis.[21]
The cause of dermatomyositis is unknown. However, genetic, immunologic, infectious, and environmental factors have been implicated.
A genetic component may predispose to dermatomyositis. Dermatomyositis rarely occurs in multiple family members, but a link to certain human leukocyte antigen (HLA) types (eg, DR3, DR5, DR7) may exist.
Polymorphisms of tumor necrosis factor (TNF) may be involved; specifically, the presence of the -308A allele is linked to photosensitivity in adults and calcinosis in children.[21, 22, 23] A meta-analysis demonstrated that the TNF-α-308A/G polymorphism might contribute to dermatomyositis susceptibility, especially in a European population.[24]
Immunologic abnormalities are common in patients with dermatomyositis. Patients frequently have circulating autoantibodies. Abnormal T-cell activity may be involved in the pathogenesis of both the skin disease and the muscle disease. In addition, family members may manifest other diseases associated with autoimmunity.
Antinuclear antibodies (ANAs) and antibodies to cytoplasmic antigens (ie, antitransfer RNA synthetases) may be present. Although their presence may help to define subtypes of dermatomyositis and polymyositis, their role in pathogenesis is uncertain.
Infectious agents have been suggested as possible triggers of dermatomyositis. These include the following:
Cases of drug-induced dermatomyositis have been reported. Dermatomyositis-like skin changes have been reported with hydroxyurea in patients with chronic myelogenous leukemia or essential thrombocytosis.[25, 26] Other agents that may trigger the disease include the following:
Dermatomyositis may be initiated or exacerbated by silicone breast implants or collagen injections, but the evidence for this is anecdotal and has not been verified in case-control studies. One report detailed HLA differences among women in whom inflammatory myopathy developed after silicone implants.[27]
The estimated incidence of dermatomyositis is 9.63 cases per million population. The estimated incidence of AMD is 2.08 cases per million.[14]
Dermatomyositis can occur in people of any age. Two peak ages of onset exist: in adults, the peak age of onset is approximately 50 years, whereas in children, the peak age is approximately 5-10 years. Dermatomyositis and polymyositis are twice as common in women as in men. Neither condition shows any racial predilection.
Most patients with dermatomyositis survive, in which case they may develop residual weakness and disability. Children with severe dermatomyositis may develop contractures. The disease may spontaneously remit in as many as 20% of affected patients. About 5% of patients have a fulminant progressive course with eventual death. Therefore, many patients require long-term therapy.
Patients with dermatomyositis who have an associated malignancy; those with cardiac, pulmonary, or esophageal involvement; and those who are elderly (ie, >60 years) have a poorer prognosis. Dermatomyositis may cause death because of muscle weakness or cardiopulmonary involvement. Patients with an associated cancer may die of the malignancy.
The association between malignancy and dermatomyositis has long been recognized. An estimated 25% of patients with dermatomyositis have or will develop an associated malignancy, and the risk appears to remain elevated for 3-5 years.[28, 29, 30] Strong data from Scandinavia, Australia, North America, and Asia continue to confirm this association with malignancy, and existing data supports that patients with CADM have a similar malignancy risk to those with classic dermatomyositis.[17, 29, 30, 31, 32, 33, 34]
Ovarian cancer is clearly over-represented in patients with dermatomyositis; however, any malignancy may occur. Reported maligancies include lung, colon, prostate, breast, pancreatic, cervical, and hematologic malignancies.[35] Predilection for certain types of malignancy may be more common in specific populations. For example, nasopharyngeal carcinoma appears to be over-represented in certain Asian populations.[36, 37, 38]
In a recently published, approximately 10-year retrospective study from southern China, 60 of 246 dermatomyositis patients developed malignancies. The risk of malignancy was highest in the first year after diagnosis of dermatomyositis, and nasopharyngeal carcinoma and ovarian carcinoma were the most common malignancies. Male gender, dysphagia and elevated erythrocyte sedimentation rate were risk factors for malignancy, whereas the presence of interstitial lung disease appeared to reduce the risk of malignancy.[39] Older age appears to be the strongest predictor of malignancy in patients with dermatomyositis.
Calcinosis may also complicate dermatomyositis. It is rare in adults but is more common in children and has been linked to delay in diagnosis and to less-aggressive therapy.[40] Contractures can occur if the patient is immobile.
Population-based studies from British Columbia concluded that patients with dermatomyositis (or polymyositis) are at increased risk for venous thromboembolism (deep venous thrombosis or pulmonary embolism)[41] and myocardial infarction,[41] especially in the first year after diagnosis. However, dermatomyositis was not associated with an increased risk of ischemic stroke.[41]
A study from Taiwan reported that the risk of osteoporosis in persons with dermatomyositis (or polymyositis) was 2.99 times higher than in those without these disorders. This risk was independent of treatment with corticosteroids and immunosuppressant drugs.[42]
African Americans and patients in lower socioeconomic groups are more likely to experience a delay in diagnosis. The prognosis in children with dermatomyositis is worse in those in whom diagnosis is delayed.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved