Chondromas are benign tumors composed of mature hyaline cartilage. They generally have limited growth potential and are not locally aggressive.[1, 2] These tumors are called enchondromas when they occur in the medullary canal of the bone[3] and are called periosteal or juxtacortical chondromas when they occur on the surface of the bone. Chondromas can also arise from the synovial sheaths of tendons or in the soft tissues adjacent to the tendons in the hand and feet of adults; in such cases, they are referred to as soft-tissue or synovial chondromas.
About 60% of enchondromas occur in the small bones of the hands and feet. The next most common sites are the long tubular bones. Of the long bones, the femur is the most commonly involved (17%), followed by the proximal humerus (7%). The proximal and distal metaphysis of the femur are involved more often than the diaphyses.
Enchondromas are rare in flat bones (eg, pelvis, ribs, scapulae, and vertebrae) and extremely rare in the craniofacial bones. Notably, they rarely affect the bones of the trunk, where chondrosarcomas are common. However, enchondromas are common in the axial skeleton, where chondrosarcomas are rare.
Periosteal chondromas typically appear on the long tubular bones, with 50% observed in the proximal humerus. The next most common sites are the short tubular bones of the hand. The usual locations of periosteal chondromas are at the tendon insertions and hence are metaphyseal; however, these tumors can develop at metaphyseodiaphyseal and diaphyseal sites as well.
NextChondromas develop from nests of growth-plate cartilage that have become entrapped in the medullary canal of the metaphysis or in the metaphyseal-diaphyseal junction. These hamartomatous proliferations persist as islands in the bone and then develop from enchondral ossification. They are subjected to the same growth factors as normal cartilage and grow until adulthood.
Chromosomal abnormalities involving the 12q13-q15 chromosomal region may be associated with chondroma, especially soft-tissue chondroma.[4] In addition, some authors have reported the involvement of chromosomes 6 and 11 in a soft-tissue chondroma.[5]
Periosteal chondroma tends to develop within and beneath the periosteal connective tissue.[6] This slow-growing tumor erodes and induces sclerosis in the contiguous cortical bone, producing the characteristic “popcorn” appearance.
Synovial chondroma results from cartilage metaplasia of subsynovial connective tissue. Karyotypic aberration has been found to be associated with synovial chondroma (ie, trisomy 5).[7]
Osteochondroma has also been associated with loss of the distal 8q chromosome. Multiple osteochondromatosis is associated with genetic defects in the 8q24.1 region (EXT1 gene) and in the 11p11-13 region (EXT2 gene).
In the United States, benign cartilage tumors account for 27.5% of all bone tumors and 70% of all cartilage tumors. Enchondromas account for 12-24% of benign bone tumors and 3-10% of all bone tumors. Periosteal chondromas account for fewer than 1% of all chondromas and 0.1% of all bone tumors. The incidence of osteochondromas is 30% of all cartilage tumors and 2% of all bone tumors. International figures for chondromas do not differ significantly from US figures.
Enchondromas are fairly evenly distributed throughout all decades of life, with no age preponderance; however, incidence peaks occur in the third and fourth decades. Periosteal chondromas are frequently diagnosed in the second or third decades of life. Osteochondromas present during adolescence and late teens. Multiple osteochondromatosis presents slightly earlier in the teens. Synovial chondromas occur from the first to the seventh decade, with a peak in the fifth decade.
No sex predilection is reported for enchondromas: the male-to-female ratio is 1:1. Similarly, there is no sex predilection for Ollier’s disease. Periosteal chondromas occur more commonly in male individuals than in female individuals. Osteochondroma has a definite male preponderance, with a ratio of 1.8:1. Synovial chondromas also show a male preponderance, with a ratio of 2:1.
Enchondromas are usually asymptomatic. In most cases, they are diagnosed incidentally during plain radiography. Expansile lesions in the short tubular bones of the hands can appear as bulbous swellings of the fingers.
Pain is a rare accompanying finding. Hence, the sudden appearance of pain in a previously asymptomatic lesion could suggest a pathologic microfracture or the transformation of a benign lesion to a malignant lesion. The rate of malignant change in solitary enchondroma is still controversial. By comparison, the rate of malignant change in Ollier disease is 10-30%, and that in Maffucci syndrome is substantially higher—as high as 100%, according to some authors.
Because periosteal chondromas are surface lesions, they may appear as palpable swellings. These tumors commonly arise where tendinous and ligamentous attachments to bone are present. The chondroma may interfere with the function of these structures and produce pain or local discomfort during activity.
The following unusual presentations of chondroma have been reported:
Complications of chondroma include the following:
In middle-aged or elderly patients, enchondroma should be differentiated from low-grade chondrosarcoma. Clinical distinctions can be made on the basis of pain, and radiologic distinctions are based on preservation of cancellous bone or adjacent cortex. In terms of histologic features, chondromas lack cellular atypia. In enchondroma, the cartilage cell nuclei are small and uniform. A homogenous chromatin is present, and multinucleated chondrocytes are infrequent. The cartilage matrix is well formed, without prominent myxoid features.
Another condition to be considered in the differential diagnosis is chondroid differentiation in fibrous dysplasia. During histologic examination, the presence of fibro-osseous elements adjacent to cartilaginous nodules and the radiologic finding of diaphyseal localization and a ground-glass appearance favor the diagnosis of fibrous dysplasia.
Bone islands and bone infarcts can mimic enchondroma. Bone islands are densely sclerotic and appear as bone within bone because of the presence of cortical bone in cancellous bone. As seen under a microscope, bone islands are composed of compact lamellar bone.
Bone infarcts commonly occur in the metaphysis of long bones around the knee. Epiphyseal involvement may be described as avascular necrosis. On radiographs, bone infarcts appear as irregular, sharply demarcated intramedullary opacities at the metaphysis or the metaphyseal-diaphyseal area. They are composed of irregular fragments of necrotic bone trabeculae, with intervening hyalinized fibrous tissue and fat necrosis. The amount of dystrophic calcification varies.
Juxtacortical chondrosarcoma may appear similar to periosteal chondroma. However, juxtacortical chondrosarcomas are usually large (>5 cm), and they do not have the buttressing of solid periosteal new bone at the margins (this lack is a characteristic feature of these lesions).
Periosteal osteosarcoma is a predominantly cartilaginous form of a surface osteosarcoma that may have the Codman triangle at its upper limits. However, periosteal osteosarcoma also lacks the buttressing of solid periosteal new bone at the margins. In periosteal chondroma, there may be excavation of the underlying cortex, but it is not associated with complete cortical disruption. Furthermore, the lesion in periosteal chondroma is clearly demarcated from the medullary cavity by a continuous rim of cortical bone.
Ollier disease is a nonhereditary skeletal disorder involving multiple enchondromas, typically metaphyseal. It commonly affects the appendicular skeleton, but the axial skeleton is not exempt. The cartilage of Ollier disease, unlike that of solitary enchondromas, has a propensity for continuous slow growth. Patients require lifelong surveillance because of a risk of secondary sarcomatous change. In some cases, surgery is indicated to correct deformity or limb-length discrepancy. In extreme cases, amputation may be needed to address stunted growth.
Maffucci syndrome consists of multiple enchondromatosis (like Ollier disease), with extraskeletal soft-tissue angiomatosis involving the skin, soft tissues, and visceral organs (including the lungs and liver). The soft-tissue angiomas are typically cavernous angiomas with frequent thrombosis and calcified thrombi.
The concern with Ollier disease and Maffucci syndrome is the incidence of secondary chondrosarcoma. Although the risk is hard to estimate for a single lesion, about 10-30% of patients with Ollier disease develop secondary chondrosarcomas. With Maffucci syndrome, at least one of the enchondromas may undergo malignant transformation at some point in the patient's lifetime.
Soft-tissue chondromas are chondromas that arise from tenosynovial sheaths or soft tissues adjacent to tendons in the hands and feet of adults. These tumors may undergo secondary changes, including dystrophic calcification, enchondral ossification, myxoid degeneration, and hemorrhage in the lesion. Because of these changes, soft-tissue chondromas may be hard to differentiate from chondrosarcomas.
Histopathologic and clinical findings should be correlated with radiographic results. Notably, even though the changes on the plain radiographs are characteristic, in 10% of synovial chondromas, no calcification can be documented in the plain radiographs. If radiography reveals a consolidated calcified mass in the joint, a synovial chondrosarcoma should be suspected.
Routine blood investigations are performed only as part of preoperative studies. Otherwise, radiologic investigations are the mainstay of the diagnosis and follow-up of chondromas.
Enchondromas produce a localized radiotranslucent defect with punctate or stippled calcifications. The amount of lucency or opacity depends on the degree of calcification. Hence, some lesions may not have radiologically visible calcification and appear lytic on radiographs, whereas other lesions may show mainly irregular calcification.
Most enchondromas are metaphyseal. This observation is consistent with the theory that these lesions are remnants of embryonic physeal cartilage that fail to ossify. Enchondromas of short tubular bones are diaphyseal, but extension to the epiphyses is frequent.
In long bones, enchondromas typically do not show distinct outlines (see the images below). This is in contrast to their appearance in short tubular bones. The contour of short tubular bones is commonly expanded, with thinning of the cortices. If a cortical breach is present along with soft-tissue extension and periosteal bone reaction, malignant transformation should be suspected.
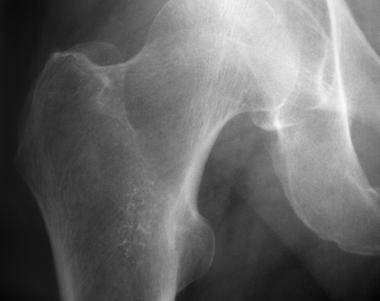 Enchondroma of proximal femur.
Enchondroma of proximal femur.
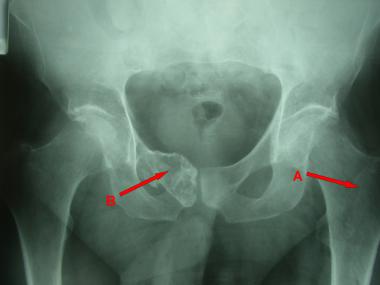 Plain radiograph reveals chondroma in left proximal femur (A) and low-grade chondrosarcoma in right superior pubic ramus and symphysis (B).
Plain radiograph reveals chondroma in left proximal femur (A) and low-grade chondrosarcoma in right superior pubic ramus and symphysis (B).
Because enchondromas are benign, their radiologic appearance should not grow or change in an adult. If a change is observed, particularly decreasing calcification or the development of translucency in the calcified lesion, chondrosarcoma or malignant transformation should be suspected.[8] The reason is that calcification occurs in resting cartilage and not in the actively dividing malignant chondrocytes of a chondrosarcoma.
The typical radiographic appearance of a periosteal chondroma is that of a well-defined surface lesion with stippled or punctate calcifications. Erosion of the underlying cortex may be noted,[9] and some long-standing tumors may be associated with subchondral sclerosis. Elevation of the periosteum overlying the lesion is a characteristic finding; on plain radiographs, this finding appears as solid buttresses of mature subperiosteal bone.
To differentiate chondromas from chondrosarcomas, clinicians should recognize that chondromas typically do not change or increase in size, shape, lucency, or calcification as seen on serial radiographs.[10]
Computed tomography (CT) is useful because it can depict subtle radiographic changes in calcification and thereby indicate early malignant transformation.
Magnetic resonance imaging (MRI) is indicated for assessing noncalcified intramedullary chondroid lesions. These tumors have low signal intensity on T1-weighted images and high signal intensity on T2-weighted images. With periosteal chondromas, MRI can show the overlying fibrous periosteum. Low signal intensity is expected on pulsed-sequence MRI images in areas where calcified chondroid matrix is demonstrated on plain radiographs.
MRI is also useful in assessing the thickness of the cartilaginous cap in osteochondroma; a cap thicker than 5 mm is grounds for concern about malignant change. Further MRI scanning is useful in assessing the soft-tissue extension and intramedullary extension in chondrosarcoma (see the image below). Dynamic contrast-enhanced (DCE) MRI appears to be useful for differentiating enchondroma from low-grade chondrosarcoma, and it has been suggested the combination of DCE-MRI with standard MRI may be more accurate for differentiating these cartilaginous tumors.[11]
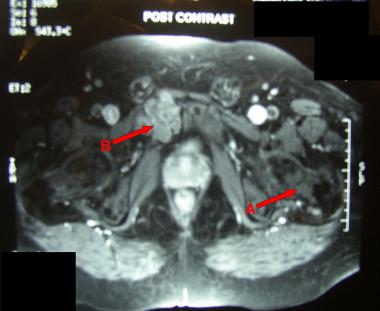 MRI showing chondroma (A) and low-grade chondrosarcoma (B).
MRI showing chondroma (A) and low-grade chondrosarcoma (B).
If the diagnosis is in doubt, biopsy is indicated. Two approaches are used.
The usual practice is to perform biopsy first and defer definitive resection. However, biopsy is not appropriate for cartilaginous lesions that are clinically and radiologically benign and is not routinely performed or advised for these lesions. Furthermore, histologic examination may not always help in correctly differentiating benign lesions from malignant ones, and it should not be relied on in isolation from the clinical and radiologic findings.
The second approach, employed in rare cases, is to plan definitive resection simultaneously with biopsy and to confirm the diagnosis by examining frozen sections.
Enchondromas are composed of subcentimeter-sized lobules.[12] Variable penetration of the lobules into the marrow produces an irregular periphery. Satellite lesions may be present. Calcifications appear ivory white on cross-section.
Periosteal chondromas appear as lobulated, cartilaginous masses with a covering of fibrous tissue from the periosteum. The inner margin produces smooth excavation in the cortex (crater effect). Thickened cortical bone clearly separates the medullary cavity of the bone.
The lesions consist of lobules of mature hyaline cartilage with varying cellularity. Cellular tumors may contain chondrocytes with plump or double nuclei. In general, the chondrocytes are similar to those seen in hyaline cartilage. Normal marrow separates the cartilaginous lobules, which may be partially encased in mature lamellar bone. Within the lesion, one may observe areas of myxoid change or focal necrosis. Foci of fine or coarse calcifications may be seen. In addition, areas of enchondral ossification may be present.
Synovial (soft-tissue) chondromas can be diffuse or localized. The former are characterized by multiple nodules that involve the entire synovium, whereas the latter are characterized by a discrete mass from a coalescence of metaplastic cartilage. In the rare nodular variety of synovial chondromatosis, a polypoidal pedunculated mass protruding into the joint may also be a feature. The large cartilaginous masses have multinodular granular surfaces, and individual nodules may range in size from less than 1 mm to 1-2 cm.
Given appropriate treatment, patients with benign chondromas generally have a good prognosis, and most remain asymptomatic.
Enchondromas of the long bones are usually asymptomatic and do not require treatment. Curettage and histopathologic evaluation are indicated if the diagnosis is uncertain, if the lesions are predominantly lytic (as seen on radiographs), if they are symptomatic and borderline in size, or if they are in any way suspicious.
Enchondromas of short tubular bones are treated with curettage and bone grafting because they tend to deform the bony contour and interfere with function. A systematic review by Bachoura et al suggested that simple curettage of hand enchondromas, without void augmentation, is safe and effective and does not increase the complication rate.[13]
Enchondromas are rare in the axial skeleton. If, however, they are found in the ribs, sternum, pelvis, or scapula, they should be treated with wide local resection and histopathologic evaluation to rule out the possibility of chondrosarcoma, which is common at these sites.
After curettage and bone grafting, enchondromas heal well with consolidation of the grafted site. If a lytic element recurs, malignancy should be ruled out. Recurrence can affect short tubular bones; it can be successfully treated with a second curettage procedure and histopathologic examination. The recurrence rate is low for surgically treated enchondromas of the hand, and the main risk factor for recurrence is a diagnosis of multiple enchondromas; malignant transformation is rare.[14]
Wide local excision is indicated for periosteal chondromas because surface chondrosarcomas can clinically and radiologically mimic these tumors. Wide local excision is a curative procedure.
Depending on the symptoms (especially if they are only mechanical), removal of loose bodies may be all that is required. Synovial chondroma is a local self-limiting condition and may reach a quiescent stage. However, if the lesion produces frequent symptoms, synovectomy may be needed to control the disease. Frequent recurrences with short intervening periods should raise the suspicion of synovial chondrosarcoma.
Chondromas that are clinically and radiographically benign should be followed up every 3-6 months and then every year only in cases where malignant degeneration is expected (as in Ollier disease or Maffucci syndrome). Otherwise, patients should be educated about the warning signs of malignant transformation (eg, deep, dull ache or night pain) and about the need for an early assessment should these signs appear.
During regular follow-up of benign chondromas, radiographs should be obtained and compared with previous images to detect any differences in the radiologic appearance of the lesion.
Patients who undergo surgical curettage and bone grafting should receive regular follow-up with radiography. Radiographs should show consolidation of the lesion and healing. Recurrence of a lytic appearance in the bone-grafted area should be treated because it suggests malignancy.
Copyright © www.orthopaedics.win Bone Health All Rights Reserved