A chondroblastoma is a rare, usually benign, tumor of bone that accounts for approximately 1% of all bone tumors. In 1931, Codman classified it as a chondromatous variant of giant cell tumors, when he described these lesions in the proximal humerus.[1] A decade later, Jaffe and Lichtenstein renamed the Codman tumor a benign chondroblastoma to emphasize the chondroblastic genesis of the lesion and to distinguish it from the classic giant cell tumor of bone.[2]
Although the exact etiology of chondroblastoma remains uncertain, the presentation, appropriate evaluation, and treatment of patients with the condition have been well described. (See Presentation, Workup, and Treatment.)
NextVarious theories have been proposed concerning the pathogenesis of chondroblastomas. Mii et al described the results of ultrastructural examination of chondroblastomas,[3] demonstrating subcellular, calcium-containing precipitates similar to those seen in chondrocytes. On the basis of these findings, the authors concluded that the tumors are of chondrogenic origin. Aigner et al, however, noted the presence of osteoid matrix–containing type I collagen and the absence of true cartilage matrix production.[4] They considered the term chondroblastoma to be a misnomer and believed that the tumor should be reclassified as a bone-forming neoplasm.
Brien et al compared the characteristics of chondroblastoma of bone to chondroblastoma of soft tissue, giant cell tumor of the tendon sheath (GCTTS), and pigmented villonodular synovitis (PVNS).[5] On examination of about 15 examples of GCTTS and PVNS, large areas of chondroid differentiation were noted that could not be distinguished from chondroblastoma of bone by either histologic or electron microscopic features. The researchers theorized that chondroblastoma of bone stems from an intraosseous proliferation of tendon sheath cells that have a predilection for chondroid formation.
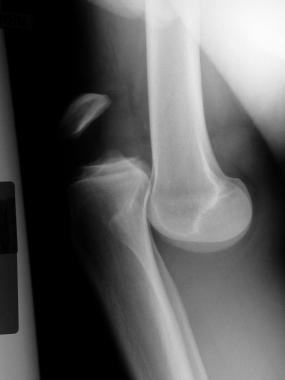
Chondroblastomas typically occur in the epiphyses of tubular long bones. The distal femoral and proximal tibial epiphyses are most frequently involved, followed by the proximal humerus, where approximately 18% of chondroblastomas appear.[6]
No risk factors are known for chondroblastoma. There have been reports of abnormalities in chromosomes 5 and 8, as well as of p53 mutations, in patients with chondroblastoma.[7] Sjögren et al performed cytogenetic analysis of benign and malignant cartilage tumors, and while they observed no consistent karyotypic abnormalities, there were recurrent breakpoints seen at 2q35, 3q21-23, and 18q21.[8]
In the United States, chondroblastoma accounts for approximately 1% of all bone tumors. The international incidence is not reported in the current literature.
Approximately 92% of patients presenting with chondroblastoma are younger than 30 years. However, chondroblastomas have been reported to arise in patients as young as 2 years and as old as 83 years. In several large series, most patients were diagnosed in the second decade of life. The male-to-female ratio is 2:1 in most series. No racial predilection is recognized.
Patients with benign chondroblastoma may limit activities due to pain. Malignant chondroblastomas, which may occur many years after the original lesion (even in the absence of radiation), are extremely rare and are associated with a dismal prognosis.
Local recurrence in long-bone lesions is approximately 10% and is higher for chondroblastomas arising in flat bones, especially those lesions arising in the vicinity of the triradiate cartilage. Average time to recurrence is 34 months after initial treatment. Most authors have not reported any significant difference in recurrence rates for tumors, regardless of the age or sex of the patient, size of the lesion, amount of calcification or vascular invasion seen on histologic examination, duration of follow-up, or method of treatment.
Springfield attributed a higher recurrence rate in patients with open physeal plates to a less aggressive curettage performed in an effort to avoid future growth arrest.[9] Recurrences may be treated with repeat curettage, with or without bone graft or cementation, and with marginal excision of any soft-tissue component.[10]
Whereas most chondroblastomas are small, well-marginated lesions that are successfully treated with intralesional curettage, a small subset of chondroblastomas behave in a much more aggressive fashion. Some of these tumors retain their benign microscopic features but nonetheless become very large or have the capability of metastasizing to the lungs and soft tissues.
Metastases may be synchronous or metachronous, occurring concurrently with the primary bone tumor or up to 33 years later. Metastases can occur even without surgical manipulation or local recurrence of the primary tumor. These more aggressive lesions may be treated with en-bloc resection and reconstruction where intralesional curettage would leave a large, bony defect. Pulmonary implants or soft-tissue metastases should be resected, especially if they are progressive.
Another rare subset of chondroblastomas may become frankly malignant even though no prior radiation therapy was used. Kyriakos et al used the term malignant chondroblastoma to describe tumors that continue to grow or disseminate, not just those that metastasize.[11] Malignant transformation typically occurs many (usually >10) years after treatment of the initial benign lesion. Pulmonary metastases may develop along with the malignant bony lesion.
Microscopic examination of the malignant bone lesion shows features similar to the original lesion (along with other areas with nuclear pleomorphism), abundant and abnormal mitotic figures, tumor necrosis, and intravascular thrombi. Ostrowski et al reported a patient with malignant transformation of a recurrent pelvic chondroblastoma with a p53 mutation.[12] Frankly malignant chondroblastoma tends to be resistant to surgery, radiation, and chemotherapy, and patients with these tumors have had dismal prognoses.
A retrospective study by Farfalli et al focused primarily on long-term joint status and functional outcomes (rather than oncologic outcomes) after curettage for epiphyseal chondroblastoma.[13] The investigators found that aggressive curettage of epiphyseal chondroblastoma frequently led to osteoarthritis and that tumors in the proximal femur appeared particularly likely to be associated with secondary osteoarthritis and prosthetic replacement.
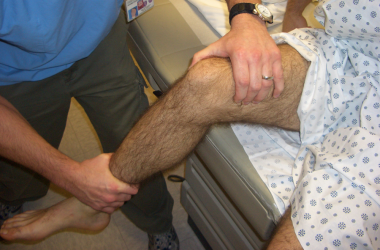
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved