Spondyloepiphyseal dysplasia (SED) is a descriptive term for a group of disorders with primary involvement of the vertebrae and epiphyseal centers resulting in a short-trunk disproportionate dwarfism. Spondylo refers to spine, epiphyseal refers to the growing ends of bones, and dysplasia refers to abnormal growth.
Two major types of SED are recognized, namely, SED congenita and SED tarda. Spranger and Wiedemann first described SED congenita in 1966,[1] and Spranger and Langer provided a further review of 29 patients in 1970.[2] In 1969, Fraser noted the particular association of SED with myopia, retinal detachment, and deafness.[3] In 1939, Jacobsen recognized SED tarda in a report of 20 patients.[4]
An image depicting spondyloepiphyseal dysplasia can be seen below.
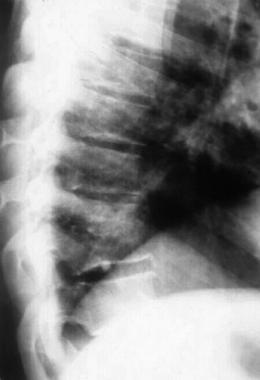
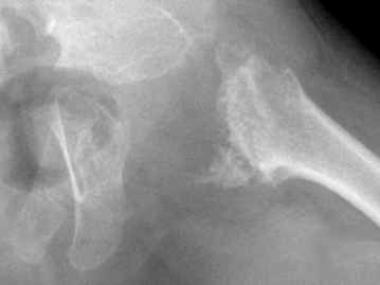 Spondyloepiphyseal dysplasia. Radiograph of the pelvis depicting delayed ossification of capital femoral epiphyses, metaphyseal flaring, horizontal acetabular roofs, triangular fragment on the inferior aspect of the broad femoral neck, and coxa vara.
Next
Spondyloepiphyseal dysplasia. Radiograph of the pelvis depicting delayed ossification of capital femoral epiphyses, metaphyseal flaring, horizontal acetabular roofs, triangular fragment on the inferior aspect of the broad femoral neck, and coxa vara.
Next
Dwarfing conditions are frequently referred to as short-limb or short-trunk types, according to whether the limbs or trunk is more extensively involved. SED, metatropic dysplasia, and Kniest syndrome are considered short-trunk dwarfing conditions. SED is a generalized dysplasia with primary involvement of the vertebrae and proximal epiphyseal centers. Other generalized dysplasias with significant vertebral involvement, such as spondylometaphyseal dysplasia or spondyloepimetaphyseal dysplasia, affect the metaphyseal region of the long bone or metaphyseal and epiphyseal region of the long bone, respectively.[5]
The clinical and radiographic differences among the various spondylodysplasias are frequently age related. SED congenita is a nonlethal form of congenital dwarfism characterized by typical skeletal dysplasias, vertebral changes, and ocular manifestations. It can be diagnosed at birth. In contrast, SED tarda is milder than SED congenita and late in onset, and appearance may be normal at birth.
With the increasing molecular definition of several types of collagen and recognition of the concentration of certain types in cartilage tissue, many skeletal dysplasias have now been defined as collagen abnormalities. Studies have indicated abnormal synthesis of type II collagen in SED congenita. Type II collagen is a primary matrix protein of physeal and epiphyseal cartilage. Because an abnormality in type II collagen should affect the molecules throughout the body, how the currently defined abnormality can translate into major structural abnormalities of the vertebrae and capital femoral epiphysis while leaving the distal femur, proximal tibia, and other regions structurally unaffected remains unclear.
Other rare forms of SED have been described. SED Maroteaux type is a form of SED with manifestations limited to the musculoskeletal system.[6] SED tarda Toledo type (first described in 1978) is a form of SED tarda with peripheral corneal opacities and a qualitative abnormality of urinary mucopolysaccharides, mainly chondroitin-6-sulfate. In 1982, Wynne-Davies recognized a form of SED tarda associated with progressive arthropathy similar to juvenile rheumatoid arthritis.[7] Kohn recognized an autosomal recessive variant of SED tarda associated with mental retardation.[8]
Similarly, other forms of SED, such as SED with brachydactyly and SED tarda Namaqualand type (NSED), have been recognized and classified under the International Nomenclature and Classification of the Osteochondrodysplasias. Bailey suggested 2 groups in addition to SED congenita and SED tarda; these are pseudo-Morquio disease and pseudoachondroplasia SED.[9] Only the common types of SED (ie, SED congenita and SED tarda) are discussed in detail in this article.[10]
International
SED congenita is a rare genetic disorder. The prevalence is approximately 3.4 per million population.[7] The incidence rate is approximately 1 per 100,000 live births.
The standardized mortality ratio is not increased for patients with SED.
Morbidity associated with SED may include the following conditions:
Most of the studies on SED involve patients from North America, Europe, and South Africa. Isolated cases have been reported from Asia and other Arab countries. Though patterns of inheritance have been identified, most cases result from sporadic mutation. No racial predilection exists.
SED congenita is autosomal dominant; hence, males and females are affected in equal numbers. Occasional cases of autosomal recessive forms have been identified.
SED tarda is X-linked recessive; hence, only males are affected. However, certain autosomal forms have been recognized, so females are occasionally affected.
SED congenita can be diagnosed at birth.
In SED tarda, appearance may be normal at birth, but the condition becomes apparent later in life. Typically, the condition is clinically manifested around puberty. However, radiographic abnormalities may appear earlier.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved