Mucopolysaccharidosis (MPS) involves defective activity of the lysosomal enzymes, which blocks degradation of mucopolysaccharides and leads to abnormal accumulation of heparan sulfate, dermatan sulfate, and keratan sulfate. MPS can be subclassified as follows:
The image below depicts Morquio syndrome.
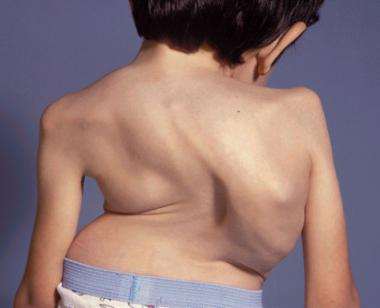 An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
Patients with MPS have normal development initially, with abnormalities appearing in infancy or later in childhood. Those with multiple organ system involvement may have the following presentations:
Findings from examination may include the following:
See Presentation for more detail.
Prenatal screening studies that may be useful include the following:
Postnatal diagnostic studies that may be helpful include the following:
Imaging studies that may be warranted are as follows:
Other tests to be considered are as follows:
See Workup for more detail.
Specific treatment or cure is limited for MPS. Management has been limited to supportive care and experimental treatment modalities. Medical treatment modalities include the following:
Surgical care for specific conditions may include the following:
Multispecialty care is mandatory for these patients and should include a pediatrician (internist), a neurologist, a cardiologist, an ophthalmologist, an audiologist, an orthopedic surgeon, and a physical and occupational therapist.
See Treatment and Medication for more detail.
NextMucopolysaccharides consist of glycosaminoglycans attached to a link protein with a hyaluronic acid core. Lysosomal enzymes degrade these macromolecules into smaller components. Heparan sulfate, dermatan sulfate, and keratan sulfate are by-products of an incomplete degradation process. The accumulation of these compounds interferes with cell function.
Different forms of mucopolysaccharidosis (MPS) were described separately throughout the 20th century. Their clinical presentations vary, depending on the type of enzyme defect and glycoprotein accumulated. (See Lysosomal Storage Disease and Madelung Deformity.)
Defective activity of the lysosomal enzymes blocks the degradation process of mucopolysaccharides, leading to abnormal accumulation of heparan sulfate, dermatan sulfate, and keratan sulfate. These degradation by-products are then secreted and detected in the urine. MPS can be subclassified according to the type and amount of substance that accumulates, as follows[1, 2, 3] :
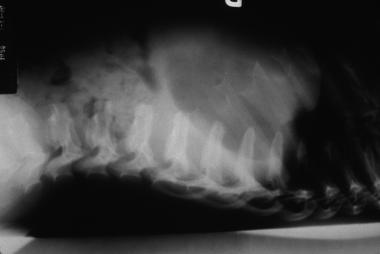 Hurler syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral plana. Courtesy of Bruce M. Rothschild, MD.
Hurler syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral plana. Courtesy of Bruce M. Rothschild, MD.
 Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
 Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
Hurler syndrome; widened metaphyses and diaphyses with truncated distal portions forming a peg characterize this radiograph. Courtesy of Bruce M. Rothschild, MD.
An 8-year-old boy with Morquio syndrome and severe kyphoscoliosis. Courtesy of Dennis P. Grogan, MD.
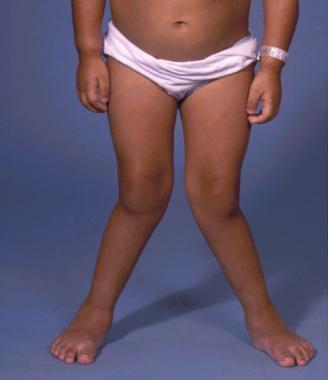 A 7-year-old girl with Morquio syndrome and typical severe genu valgum. Courtesy of Dennis P. Grogan, MD.
A 7-year-old girl with Morquio syndrome and typical severe genu valgum. Courtesy of Dennis P. Grogan, MD.
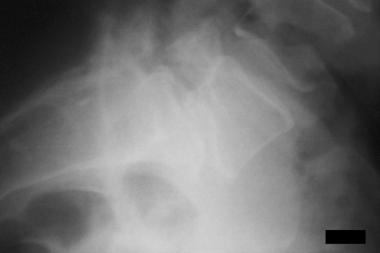
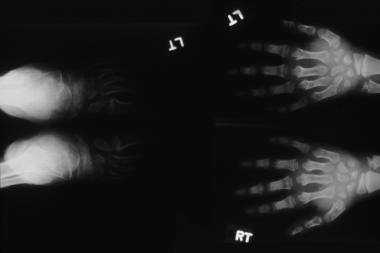
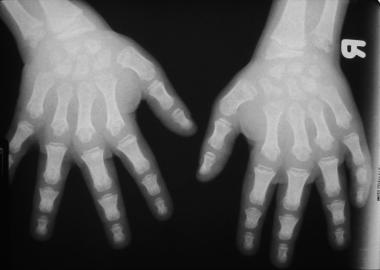 Morquio syndrome; widened bases of phalanges with osteopenia. Courtesy of Bruce M. Rothschild, MD.
Morquio syndrome; widened bases of phalanges with osteopenia. Courtesy of Bruce M. Rothschild, MD.
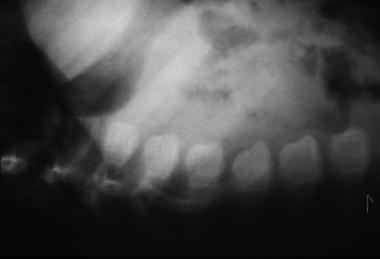 Morquio syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral body beaking. Courtesy of Bruce M. Rothschild, MD.
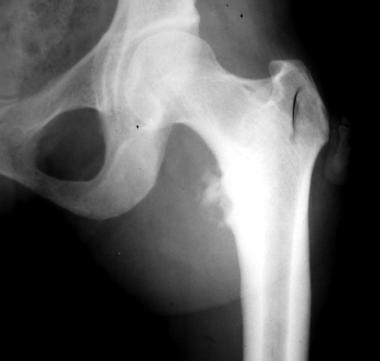
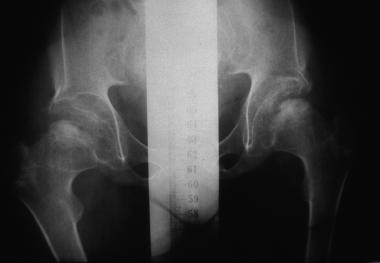
Morquio syndrome; anteroposterior radiograph of pelvis illustrates avascular necrosis of femoral head. Courtesy of Bruce M. Rothschild, MD.
Morquio syndrome; lateral radiograph of thoracolumbar vertebrae illustrates vertebral body beaking. Courtesy of Bruce M. Rothschild, MD.
Morquio syndrome; anteroposterior radiograph of pelvis illustrates avascular necrosis of femoral head. Courtesy of Bruce M. Rothschild, MD.
Defective activity of the lysosomal enzymes blocks the degradation process of mucopolysaccharides, leading to abnormal accumulation of heparan sulfate, dermatan sulfate, and keratan sulfate.
All forms of MPS are inherited as autosomal recessive disorders, with the exception of Hunter syndrome (MPS II), which is inherited as sex-linked recessive.
Worldwide, the prevalence of all types of MPS is 1 case in 16,000-30,000 births. MPS III accounts for 80% of cases.
Ages at which features of MPS present are somewhat variable. MPS features mostly present in the first few months of life. However, Morquio syndrome usually presents in children aged 2-4 years, and MPS IS and MPS VI can present late in childhood.
All mucopolysaccharidoses are inherited as autosomal recessive disorders with the exception of Hunter syndrome (MPS II), which is inherited as sex-linked recessive condition; thus, all patients with Hunter syndrome are males.
These syndromes are found in all ethnic groups. Incidence of MPS II is increased in Israeli Jews, and incidence of MPS IV is increased in French Canadians.
The prognosis varies depending on the type of MPS. Most of these patients have shortened life spans, and some of them die in infancy. These disease processes have significant effects on the growth and development of the musculoskeletal system, including joint stiffness or hyperlaxity, deformities, and progressive loss of function. Multiple other organ systems are involved. The type and extent of organ system involvement are variable depending on the subset of the disease.
Bone marrow transplantation has some positive effects systemically, such as reduction in hepatosplenomegaly, airway obstruction, and cardiopulmonary disease. These effects have resulted in improved life span, and many of these patients survive beyond the first decade of life.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved