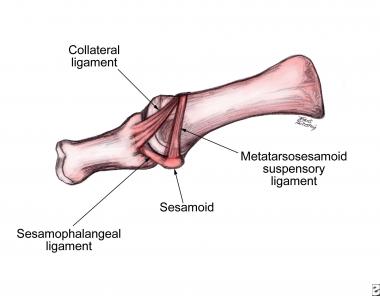
Skeletal dysplasias are a heterogeneous group of dysplasias that include more than 200 recognized conditions. They are disorders of growth and remodeling of bone and cartilage. Most disorders result in short stature, which is defined as height more than two standard deviations below the mean for the population at a given age. (When one discusses height in patients with short stature, one may say "smaller than average" rather than "dwarf.")
Lamy and Maroteaux first delineated this syndrome in 1960 and coined the term diastrophic dwarfism.[1] The term diastrophic is derived from the Greek word diastrophe ("distortion, twisting"); it is a geologic term used to describe the bending and twisting of the earth's crust during geomorphogenesis. This name seems appropriate for this disorder, in which the skeleton appears twisted.
In 1977, at the Second International Conference for Nomenclature for Constitutional Diseases of Bone, the name was changed from diastrophic dwarfism to diastrophic dysplasia.[2, 3] The term pseudodiastrophic dwarfism is used for a disorder that is clinically, radiologically, and histologically distinct from true diastrophic dysplasia, and it should not be used inadvertently.
Conditions that cause dwarfing are frequently referred to as short-limb or short-trunk types, depending on whether the trunk or limbs are more extensively involved. Diastrophic dysplasia is considered a short-limb dwarfing condition. Additional terms used to describe the segment of the limb with the greatest involvement are rhizomelic (proximal), mesomelic (middle), and acromelic (distal). In diastrophic dysplasia, the extremity involvement is rhizomelic (in 40% of cases) or mesomelic (in 29% of cases).
Diastrophic dysplasia[4] is a recessively inherited chondrodysplasia, one of which is particularly common in Finland. This term describes dwarfism with perhaps the most numerous and severe skeletal abnormalities from cervical spine to the feet. In the past, this condition was referred to as achondroplasia with clubfeet or arthrogryposis multiplex congenita.
A distinct group of patients who have many features of diastrophic dysplasia are referred to as having diastrophic variants; these individuals are taller and less severely affected than persons with classic diastrophic dwarfism. Classic diastrophic dysplasia and diastrophic variants are different expressions of a single genetic disorder (with variable penetrance) rather than separate entities. Individuals identified as having a diastrophic variant should be referred to as having mild diastrophic dysplasia.
Impairment of physeal, epiphyseal, and articular cartilage throughout the body is responsible for characteristic findings. Unlike those with achondroplasia or hypochondroplasia, patients with diastrophic dysplasia have epiphyseal involvement and are at risk for degenerative joint disease. Although the development and growth of cartilaginous structures are disturbed, the intramembranous ossification and appositional growth pattern are not primarily affected.
NextProteoglycans are considered to be among the chief constituents of cartilage. Undersulfation of proteoglycan in the cartilaginous matrix is responsible for the impairment of performance and load-bearing ability of physeal, epiphyseal, and articular cartilage throughout the body.
The DTDST protein acts as a sodium-independent sulfate/chloride transporter and belongs to the SLC26 anion transporter family. The Finnish founder mutation in DTDST has been identified.[5] Approximately 95% of affected Finnish patients have a rare ancestral haplotype that was found in only 4% of a Finnish control population. The founder mutation is a guanine-thymine (GT) to guanine-cytosine (GC) transition in the splice donor site of a 5'-untranslated exon of the DTDST gene. The mutation acts by severely reducing levels of mRNA of the DTDST transcript.[6]
Mutations in DTDST are responsible for a family of chondrodysplasias that include four recessively inherited conditions: diastrophic dysplasia, multiple epiphyseal dysplasia, atelosteogenesis type 2, and achondrogenesis type 1B. In addition to the intrinsic sulfate transport properties of the DTDST protein, other factors influence the phenotype in individuals with these mutated alleles.[7]
In 1994, Hastbacka et al identified the gene DTDST. This gene, which codes for a sulfate transporter protein, has been mapped to distal end of chromosome bands 5q31-q34.[5] DTDST is inherited in an autosomal recessive manner.[8] Diastrophic dysplasia and McKusick-type metaphyseal chondrodysplasia are the only skeletal dysplasias with autosomal recessive transmission.
About 5% of cases may involve sporadic new mutations. Both parents of an affected individual are carriers of the abnormal gene but are clinically healthy. For a carrier couple, each pregnancy entails a 25% risk of producing an affected child. Each unaffected full sibling of an affected individual has a 67% likelihood of being a carrier. The offspring of an affected individual is a carrier and therefore unaffected unless the other parent is a carrier or affected with the same condition.
Diastrophic dysplasia is a disorder with a wide range of clinical manifestations; this variation has important implications. Parents of a child with mild diastrophic dysplasia, which would previously have been called a diastrophic variant, must be informed that they are at 25% risk of having other children with disproportionate dwarfism. In addition, they should be made aware that the expression of the disorder may be more severe in subsequent children.
Although diastrophic dysplasia is extremely rare, the percentage of carriers in certain groups is high. In Finland, 1-2% of the general population are carriers, and a total of 183 cases have been diagnosed, with a prevalence of 1 per 30,000 population. Diastrophic dysplasia has been observed in most white populations.[9]
Diastrophic dysplasia is an autosomal recessive disorder and occurs with equal frequency in males and females.
Patients have a minimally (5%) increased rate of perinatal mortality due to cervical myelopathy or respiratory problems such as aspiration pneumonia and laryngotracheomalacia. Patients with severe spinal deformities are also predisposed to the development of respiratory problems. A lethal form of diastrophic dysplasia has been described that can cause death soon after birth due to cardiorespiratory insufficiency. Overall, life expectancy is not reduced, and patients are able to lead productive lives at work and with their families.
Patients may benefit from the information on the Web site for diastrophic dysplasia, Diastrophic Help.
An important resource for individuals with short stature is the Little People of America (LPA) Organization. The LPA is a national organization that addresses the social, physical, and medical needs of its constituency. It holds annual regional and national conventions. Philosophically, this organization emphasizes the positive aspects of their members' abilities and lives rather than presenting short stature as a disability.
The Dwarf Athletic Association of America (DAAA) is a member of the US Olympic Committee that promotes athletic participation for individuals with short stature.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved